scanpy单细胞转录组python教程(四):单样本数据分析之降维聚类及细胞注释
接上节(scanpy单细胞转录组python教程(一):不同形式数据读取,scanpy单细胞转录组python教程(二):单样本数据分析之数据质控,scanpy单细胞转录组python教程(三):单样本数据分析之数据标准化、特征选择、细胞周期计算、回归等)。这一节是scanpy单细胞分析流程的最后一个内容了,完成降维聚类及细胞注释,这里我们还是推荐使用marker手动注释,自动注释这里先不演示。完成细胞注释,获得的结果是后续所有分析的基石。
首先加载数据:
import pandas as pd
import scanpy as sc
sc.settings.verbosity = 3
sc.settings.set_figure_params(dpi=80, facecolor="white")
adata = sc.read_h5ad('./adata.h5ad')
adata
# AnnData object with n_obs × n_vars = 8419 × 22022
# obs: 'n_genes', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_20_genes', 'pct_counts_mt', 'pct_counts_ribo', 'pct_counts_hb', 'S_score', 'G2M_score', 'phase'
# var: 'gene_ids', 'feature_types', 'n_cells', 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
# uns: 'hvg', 'log1p', 'pca', 'phase_colors'
# obsm: 'X_pca'
# varm: 'PCs'
# layers: 'counts', 'log1p_norm'PCA降维:通过运行主成分分析(PCA)来降低数据的维度。运行结束后可以使用sc.pl.pca_variance_ratio函数展示PC,默认展示前30个PCs,来看看单个PC分量对数据总方差的贡献情况,这个类似于seurat中的Eblowplot。以便让我们在后续的分析中选择合适的PCs。PCs的选择实际中可能需要自行确定调整。
sc.tl.pca(adata, svd_solver="arpack",use_highly_variable=True)
sc.pl.pca_variance_ratio(adata, log=False,n_pcs=50)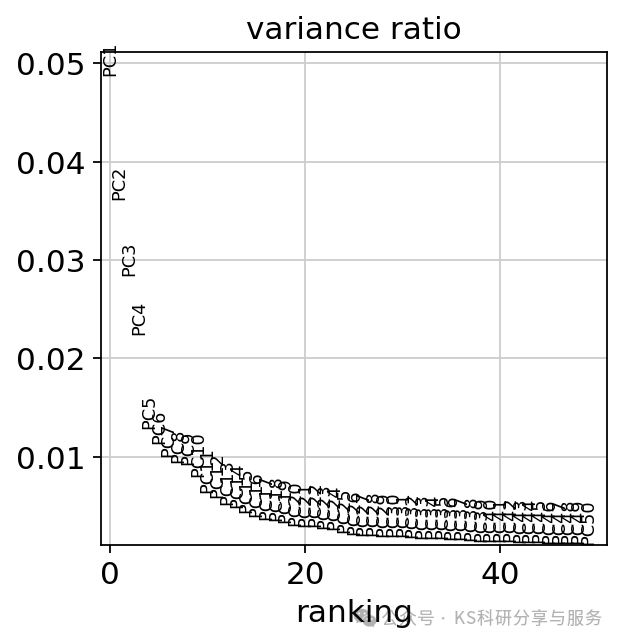
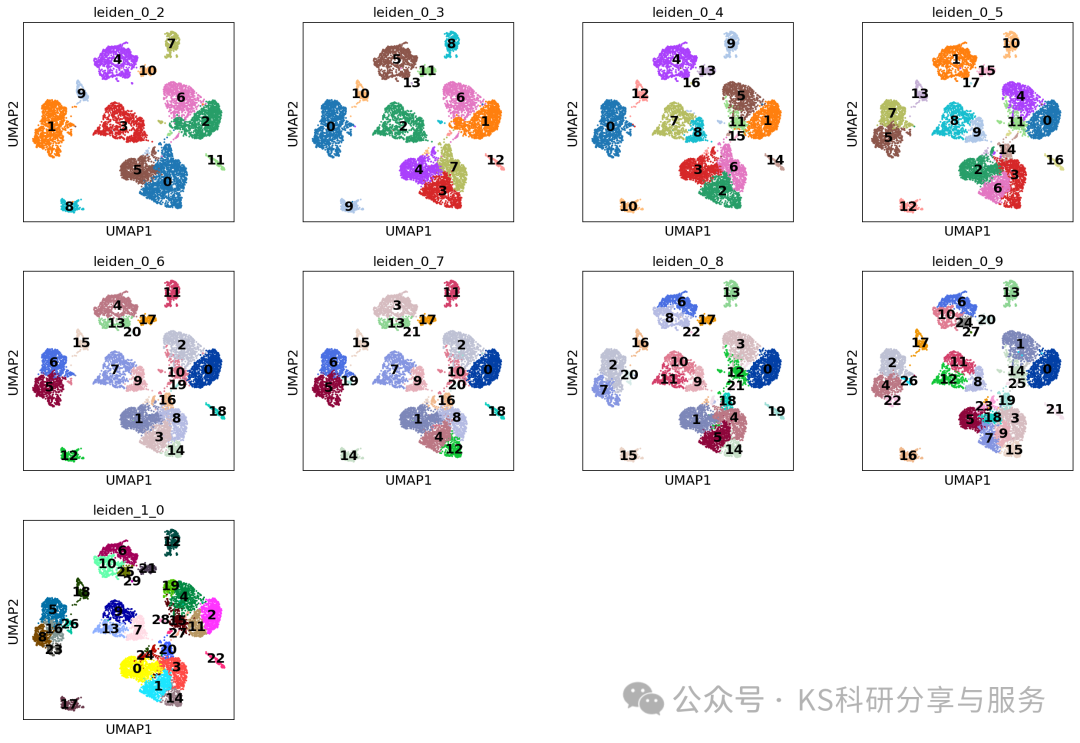
Clustering:使用scanpy函数sc.pp.neighbors细胞的邻域图。这里我们选择前20个主成分调用此函数,这些主成分能够捕捉到数据集中的大部分变异。
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=20)Clustering the neighborhood graph scanpy 中,默认的分辨率参数为1.0。但在许多情况下,我们可能希望尝试不同的分辨率参数来控制聚类的粗细程度。因此,可以将聚类结果保存在指定的key,用于指示所选的分辨率。通常可以多选择几个分辨率,类似于seurat,一般0.2-1.2就足够获取合适的结果。
# sc.tl.leiden(
# adata,
# resolution=0.9,
# random_state=0,
# flavor="igraph",
# n_iterations=2,
# directed=False,)#首次运行可以出现这个error,Please install the igraph package: `conda install -c conda-forge python-igraph` or `pip3 install igraph`.
#ImportError: Please install the leiden algorithm: `conda install -c conda-forge leidenalg` or `pip3 install leidenalg`.
#安装igraph即可: pip install igraph -i https://pypi.tuna.tsinghua.edu.cn/simple
#pip install leidenalg -i https://pypi.tuna.tsinghua.edu.cn/simple
list_leiden_res = [0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0]
for leiden_res in list_leiden_res:print(leiden_res)sc.tl.leiden(adata, resolution=leiden_res, key_added='leiden_'+str(leiden_res).replace(".", "_"))# 0.2
# running Leiden clustering
# finished: found 12 clusters and added
# 'leiden_0_2', the cluster labels (adata.obs, categorical) (0:00:03)
# 0.3
# running Leiden clustering
# finished: found 14 clusters and added
# 'leiden_0_3', the cluster labels (adata.obs, categorical) (0:00:02)
# 0.4
# running Leiden clustering
# finished: found 17 clusters and added
# 'leiden_0_4', the cluster labels (adata.obs, categorical) (0:00:03)
# 0.5
# running Leiden clustering
# finished: found 18 clusters and added
# 'leiden_0_5', the cluster labels (adata.obs, categorical) (0:00:02)
# 0.6
# running Leiden clustering
# finished: found 21 clusters and added
# 'leiden_0_6', the cluster labels (adata.obs, categorical) (0:00:02)
# 0.7
# running Leiden clustering
# finished: found 22 clusters and added
# 'leiden_0_7', the cluster labels (adata.obs, categorical) (0:00:02)
# 0.8
# running Leiden clustering
# finished: found 23 clusters and added
# 'leiden_0_8', the cluster labels (adata.obs, categorical) (0:00:02)
# 0.9
# running Leiden clustering
# finished: found 28 clusters and added
# 'leiden_0_9', the cluster labels (adata.obs, categorical) (0:00:02)
# 1.0
# running Leiden clustering
# finished: found 30 clusters and added
# 'leiden_1_0', the cluster labels (adata.obs, categorical) (0:00:03)
list_leiden_res = [0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0]
resolutions = ['leiden_' + str(res).replace(".", "_") for res in list_leiden_res]
resolutions
#可视化所有分辨率下的结果
sc.pl.umap(adata,color= resolutions,legend_loc="on data",
)
Finding marker genes:鉴定marker基因,用于后续的分群注释。前面的res选择一个差不多合适的即可,对于大分群,也没必要一次性选择很大的res。
sc.tl.rank_genes_groups(adata, groupby = "leiden_0_3", method="wilcoxon",layer='log1p_norm')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)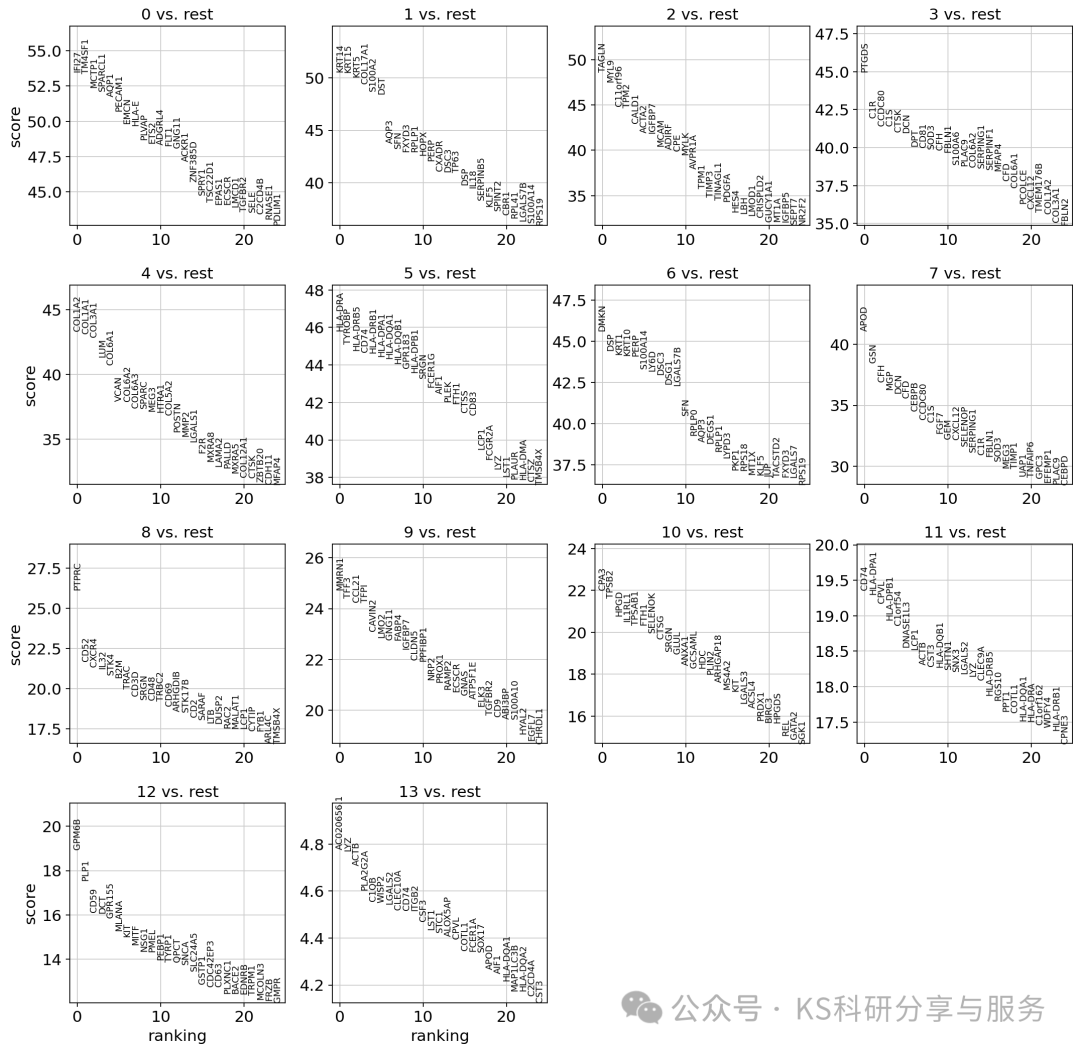
# 将结果转换为 DataFrame.
marker_genes_df = sc.get.rank_genes_groups_df(adata, group=None, key='rank_genes_groups')
print(marker_genes_df.head())
#筛选显著性的结果
significant_markers = marker_genes_df[(marker_genes_df['pvals_adj'] < 0.05) & (marker_genes_df['logfoldchanges'] > 1)]
significant_markerscell annotation:细胞的注释我还是倾向于手动注释,使用marker基因。使用经典的marker,dotplot看看cluster表达情况。
marker_genes = [*["KRT5", "KRT10", "KRT14", "KRT15"], *["THY1", "COL1A1", "COL11A1"], *["CD3D", "CD8A", "CD4","IKZF2", "CCL5"], *["CD14", "CD86", "CD163", "CD1A", "CLEC9A", "XCR1"], *["MITF", "SOX10", "MLANA"], *["VWF", "PECAM1", "SELE"], *["FLT4", "LYVE1"], *["TPM1", "TAGLN", "MYL9"], *["TRPC6", "CCL19"], *["KIT", "TPSB2", "HPGD"], *["ITGB8", "CD200", "SOX9"]]
sc.pl.dotplot(adata, marker_genes, groupby="leiden_0_3",dendrogram=True);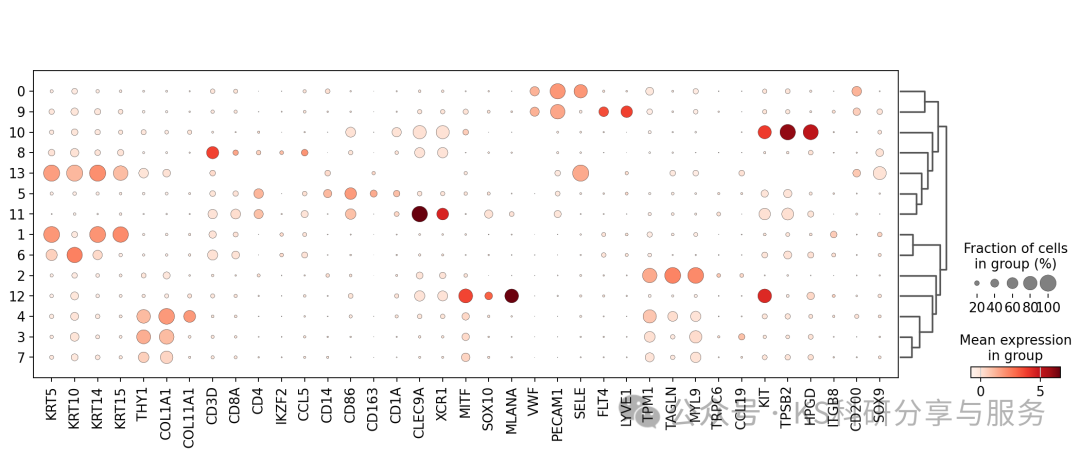
#注释celltype
cellAnno = {"0": "Endothlial","1": "Kers","2": "MuCs","3": "Fibs","4": "Fibs","5": "APCs","6": "Kers","7": "Fibs","8": "Tcells","9": "Lym","10":"Mast","11":"APCs","12":"Mela","13":"lowQ"}
#将上面的注释字典与adata中的leiden_0_3 match,并且形成新的一列obs celltype
adata.obs["celltype"] = adata.obs.leiden_0_3.map(cellAnno)
adata
# AnnData object with n_obs × n_vars = 8419 × 22022
# obs: 'n_genes', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_20_genes', 'pct_counts_mt', 'pct_counts_ribo', 'pct_counts_hb', 'S_score', 'G2M_score', 'phase', 'leiden_res0_3', 'leiden_res0_5', 'leiden_res1', 'leiden_0_2', 'leiden_0_3', 'leiden_0_4', 'leiden_0_5', 'leiden_0_6', 'leiden_0_7', 'leiden_0_8', 'leiden_0_9', 'leiden_1_0', 'celltype'
# var: 'gene_ids', 'feature_types', 'n_cells', 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
# uns: 'hvg', 'log1p', 'pca', 'phase_colors', 'neighbors', 'umap', 'leiden_res0_3', 'leiden_res0_5', 'leiden_res1', 'leiden_res0_3_colors', 'leiden_res0_5_colors', 'leiden_res1_colors', 'leiden_0_2', 'leiden_0_3', 'leiden_0_4', 'leiden_0_5', 'leiden_0_6', 'leiden_0_7', 'leiden_0_8', 'leiden_0_9', 'leiden_1_0', 'leiden_0_2_colors', 'leiden_0_3_colors', 'leiden_0_4_colors', 'leiden_0_5_colors', 'leiden_0_6_colors', 'leiden_0_7_colors', 'leiden_0_8_colors', 'leiden_0_9_colors', 'leiden_1_0_colors', 'rank_genes_groups', 'dendrogram_leiden_0_3'
# obsm: 'X_pca', 'X_umap'
# varm: 'PCs'
# layers: 'counts', 'log1p_norm'
# obsp: 'distances', 'connectivities'
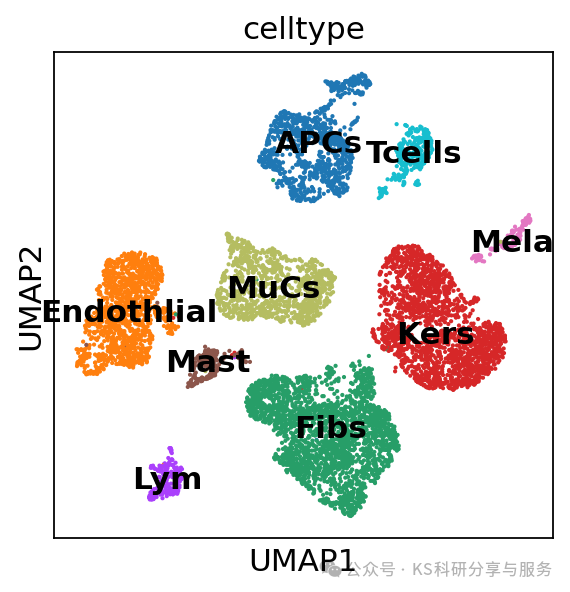
sc.pl.umap(adata, color=["celltype"])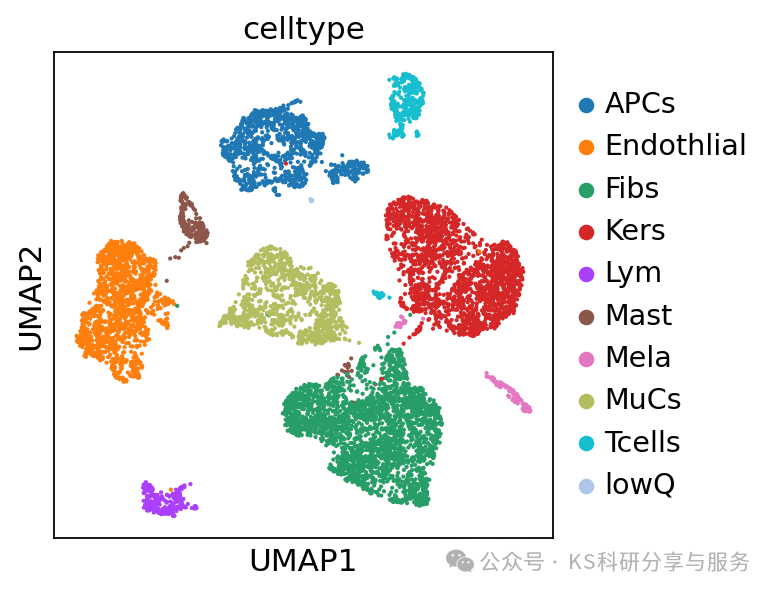
#lowQ这群只有8个细胞,质量不好,所以我们决定将这群细胞删除,在删除前,将原来的数据备份
adata_all = adata.copy()
adata_filtered = adata[~adata.obs['celltype'].isin(['lowQ']), :].copy()
adata_filtered
sc.pl.umap(adata_filtered,color= 'celltype',legend_loc="on data",
)删除细胞后,需要重新计算邻居图等依赖细胞数的结构,运行neighbors and umap!sc.pp.neighbors(adata_filtered, n_neighbors=10, n_pcs=20)
sc.tl.umap(adata_filtered)
sc.pl.umap(adata_filtered,color= 'celltype',legend_loc="on data",
)