Melon: 基于marker基因的三代宏基因组分类和定量软件

基于标志基因的分类方法在理论上(如生物学意义)和实际应用中(如计算资源消耗)均具有优势。除了我们了解的可能的假阴性,三代宏基因组的类似分析工具比较欠缺,于是作者就开发了一个。
来自港大张彤团队去年的力作,得知这个软件可能最早是在团队的主页,但是真正感兴趣是看到NM的三代宏基因组扩展陆地微生物的研究中使用了这个软件。虽然文章发表的期刊不是特别棒,但看到最近两个月依然在更新,想要尝试下分析的效果啦!
软件安装和数据库准备
软件名就是取自文章标题的缩写的前几个字母,这也是一般软件起名的通用做法啦,瓜,名字还挺有意思的!安装就是大家还主流使用的conda啦,新建个环境,然后激活。数据库准备,还是挺耗费时间和空间的,不过一般基于marker基因的宏基因组数据库,体积小很多啦!下载后使用三代常用的minimap建立索引。
如果宿主含量特别高,并且想要估计原核生物的平均基因组大小,需要通过适当的比对或启用简单的预过滤模块来去除它们。如kraken数据库的不同版本:PlusPF,标准+原生+真菌,PlusPFP,多了个植物,可以使用限制数据库大小为8G的等小体积版本。
# 安装
conda create -n melon -c conda-forge -c bioconda melon
conda activate melon
# 数据库,想要用的新的库,用0.3版本
## NCBI 3.2G
wget -qN --show-progress https://zenodo.org/records/15231351/files/database.tar.gz
tar -zxvf database.tar.gz
## GTDB
# wget -qN --show-progress https://zenodo.org/records/15231379/files/database.tar.gz
# tar -zxvf database.tar.gz
# 建索引
## if you encounter memory issue please consider manually lowering cpu_count or simply set cpu_count=1
cpu_count=$(python -c 'import os; print(os.cpu_count())')diamond makedb --in database/prot.fa --db database/prot --quiet
ls database/nucl.*.fa | sort | xargs -P $cpu_count -I {} bash -c 'filename=${1%.fa*};filename=${filename##*/};minimap2 -x map-ont -d database/$filename.mmi ${1} 2> /dev/null;echo "Indexed <database/$filename.fa>.";' - {}## remove unnecessary files to save space
rm -rf database/*.fa
# kraken https://benlangmead.github.io/aws-indexes/k2
mkdir database_kraken
wget -q --show-progress https://genome-idx.s3.amazonaws.com/kraken/k2_pluspf_08gb_20230605.tar.gz -O database_kraken/db.tar.gz
tar -zxvf database_kraken/db.tar.gz -C database_kraken## remove temporary files to save space
rm -rf database_kraken/db.tar.gz
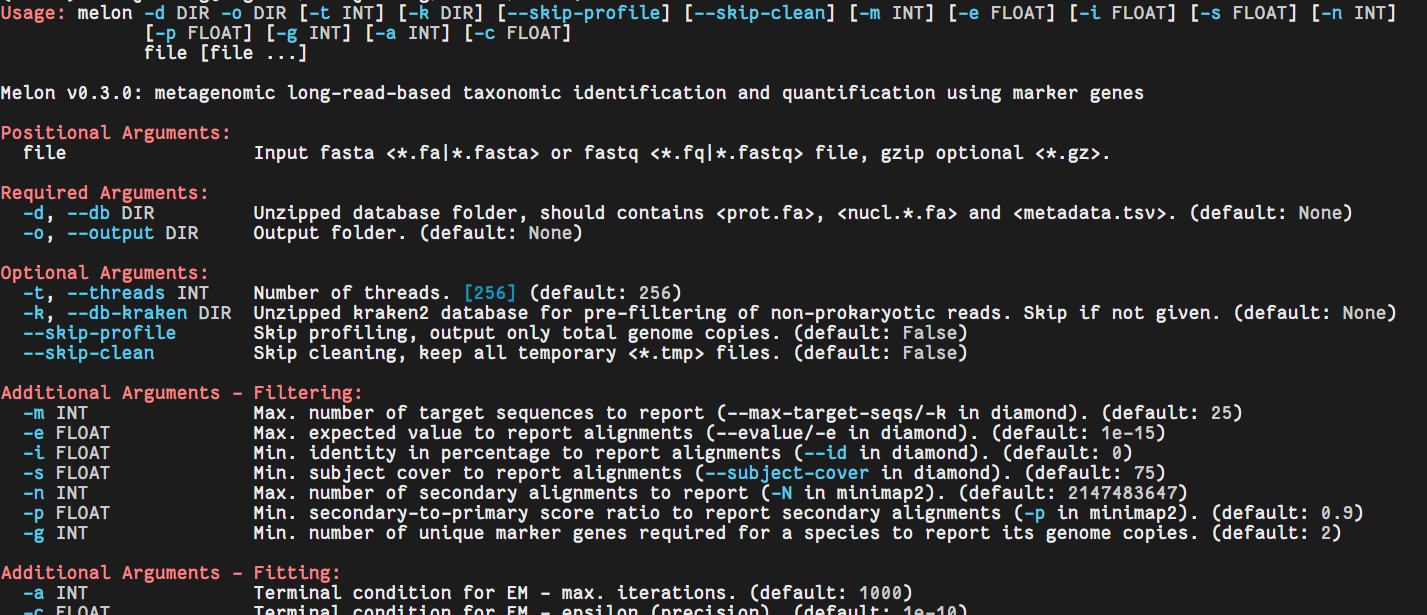
melon -h
软件的使用说明相对简洁,并没有提供详细的说明文档,只有参数的提示。可以看到必须参数就是数据库-d和输出文件夹-o,输入支持fasta或fastq,以及压缩后的,常规操作习惯啦。
可选参数有-t是线程数,默认竟然是256,-k是指定kraken数据库作过滤。–skip-profile跳过丰度分析,只分析基因组拷贝数,–skip-clean保留中间文件。
然后就是blast/diamond的一些过滤参数啦,-m指定命中候选数量,-e就是evalue啦,-i是相似度,-s是报告比对结果的最小目标覆盖百分比。
接着是minimap2的过滤参数,-n是控制每个查询序列最多输出多少个次级比对。-p是控制是否输出次级比对(secondary mappings),依据其得分与主比对(primary mapping)的相对比例。
其他参数还有-g是一个物种报告其基因组拷贝所需的最少独特标记基因数量。最后就是拟合的两个附加参数,-a 期望最大化算法的终止条件 - 最大迭代次数。-c 期望最大化算法的终止条件 - 精度(ε)。
当一个序列可以在多个位置比对时,除了主比对(primary alignment),Minimap2 还可以输出若干个得分较高的次级比对。这个参数可以限制这些次级比对的数量,以避免输出过多冗余信息
使用
建议使用质控后的序列作为输入,比如ONT常用的质控指标最小质量得分10;最小读取长度1,000 bp(nanoq -q 10 -l 1000)。运行命令是比较简单的,参数也比较少,就指定下数据库和输出就可以啦。
# 如果过滤非原核
melon *.fa -d database -o . -k database_kraken
# 不过滤直接运行的demo
wget -qN --show-progress https://zenodo.org/records/12571849/files/example.fa.gz
melon example.fa.gz -d database -o .
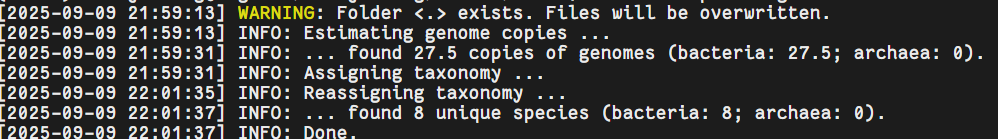
结果有两个,一个是json格式,另一个是tsv,一般咱看tsv啦!从门到属的信息以及应该是taxid,拷贝数,丰度和相似度。
| superkingdom | phylum | class | order | family | genus | species | copy | abundance | identity |
|---|---|---|---|---|---|---|---|---|---|
| 2|Bacteria | 1224|Pseudomonadota | 1236|Gammaproteobacteria | 72274|Pseudomonadales | 135621|Pseudomonadaceae | 286|Pseudomonas287|Pseudomonas | aeruginosa | 2.125 | 7.727273e-02 | 0.9571/0.9473 |
好啦,就分享到这,期待大家的真实项目使用体会,欢迎交流!
参考
- Chen, X., Yin, X., Shi, X. et al. Melon: metagenomic long-read-based taxonomic identification and quantification using marker genes. Genome Biol 25, 226 (2024). https://doi.org/10.1186/s13059-024-03363-y
- https://github.com/xinehc/melon