单细胞数据分析:单细胞计数矩阵(Seurat)
在使用seurat进行单细胞分析的时候,大多数的教程都是用计数矩阵作为数据输入,但是我发现一些新手朋友对于不同数据库来源(GEO、BD)的数据或者想要去复现、借鉴一个感兴趣的文章中的下机数据时,不知道怎么把数据处理成Seurat可以读入的计数矩阵,所以本篇文章就详细介绍单细胞数据的上游分析。
10X平台数据
上游分析主要涉及的步骤就两个:比对和质控。我们可以先从10X平台官网了解一些软件和方法。
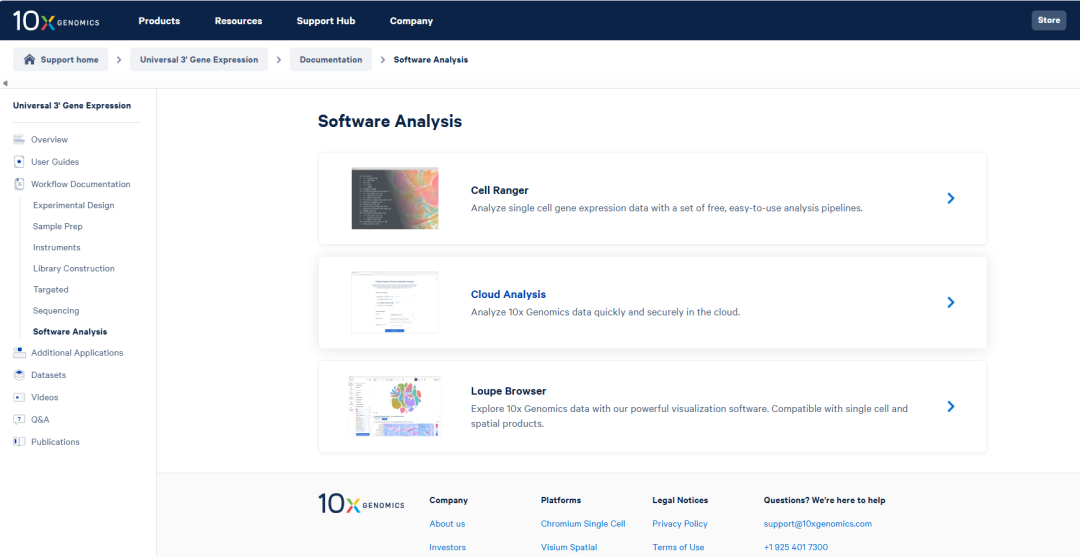
“Software Analysis”的界面提供了3种工具:
Cell Ranger:比对质控需要用到的软件
Cloud Analysis:在线云分析软件,提供fastq文件即可分析
Loupe Browser:比对质控后结果的初步可视化
我们今天重点学Cell Ranger的使用。
Cell Ranger 配置和使用
官网下载:
# 使用curl命令下载
curl -o cellranger-8.0.0.tar.gz "https://cf.10xgenomics.com/releases/cell-exp/cellranger-8.0.0.tar.gz?Expires=1712352870&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA&Signature=lAp0t6wo-oNLmAhcfNGu6Nqie6OOcWcWoZWx9Lj2JxE8Gj1Do8p2rVYH~PE4f0wCemkrQKilRUDa42FRKskUugvKT59MrZDj9fkWy4lxlPlMOBFOLJG~eZOQJxwKgTzkXdYGuBr~J~k0KGTF0rCYBH0p6s5BB5Bff5Rx033~sXxr-YU8tCgHtx7YZOg67ov-NEMqh3y-S~CcyRV2RcQkv1nrcFMrLoC4SxoV8U4LjlzGqTPxFzSMZ2-xQYOs5wWw0N2TnrlhV~ofY6xHvPmSfEzhnWt3UHBTc5zjBSoP3bKpsFaFWFInyrZXUMILV6iXzeiEvp4T7hq5g9S2TfmaUQ__"# 使用wget命令下载
wget -O cellranger-8.0.0.tar.gz "https://cf.10xgenomics.com/releases/cell-exp/cellranger-8.0.0.tar.gz?Expires=1712352870&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA&Signature=lAp0t6wo-oNLmAhcfNGu6Nqie6OOcWcWoZWx9Lj2JxE8Gj1Do8p2rVYH~PE4f0wCemkrQKilRUDa42FRKskUugvKT59MrZDj9fkWy4lxlPlMOBFOLJG~eZOQJxwKgTzkXdYGuBr~J~k0KGTF0rCYBH0p6s5BB5Bff5Rx033~sXxr-YU8tCgHtx7YZOg67ov-NEMqh3y-S~CcyRV2RcQkv1nrcFMrLoC4SxoV8U4LjlzGqTPxFzSMZ2-xQYOs5wWw0N2TnrlhV~ofY6xHvPmSfEzhnWt3UHBTc5zjBSoP3bKpsFaFWFInyrZXUMILV6iXzeiEvp4T7hq5g9S2TfmaUQ__"解压配置:
# 解压下载的压缩包
tar -zxvf cellranger-8.0.0.tar.gz# 进入解压后的文件夹中,然后输入下面的命令,将cellranger的配置写进bashrc中
echo "export PATH=${PWD}:\$PATH" >> ~/.bashrc# 然后调用cellranger空运行,弹出了帮助文档,说明安装成功
$ cellranger## cellranger cellranger-8.0.0
##
## Process 10x Genomics Gene Expression, Feature Barcode, and Immune Profiling data
##
## Usage: cellranger
##
## Commands:
## count Count gene expression and/or feature barcode reads from a single sample and GEM well## multi Analyze multiplexed data or combined gene expression/immune profiling/feature barcode data## multi-template Output a multi config CSV template## vdj Assembles single-cell VDJ receptor sequences from 10x Immune Profiling libraries## aggr Aggregate data from multiple Cell Ranger runs## reanalyze Re-run secondary analysis (dimensionality reduction, clustering, etc)## mkvdjref Prepare a reference for use with CellRanger VDJ## mkfastq Run Illumina demultiplexer on sample sheets that contain 10x-specific sample index sets## testrun Execute the 'count' pipeline on a small test dataset## mat2csv Convert a feature-barcode matrix to CSV format## mkref Prepare a reference for use with 10x analysis software. Requires a GTF and FASTA## mkgtf Filter a GTF file by attribute prior to creating a 10x reference
## upload Upload analysis logs to 10x Genomics support
## sitecheck Collect Linux system configuration information
## help Print this message or the help of the given subcommand(s)##
## Options:
## -h, --help Print help## -V, --version Print version参考基因组文件的下载:
选择下载依据人或小鼠的参考基因组创建的比对索引文件
# 使用curl命令下载
curl -O "https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCm39-2024-A.tar.gz"
# 使用wget命令下载
wget "https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCm39-2024-A.tar.gz"
# 下载好之后解压
tar -zxvf refdata-gex-GRCm39-2024-A.tar.gz我选择了一个成年小鼠心脏的数据,细胞数大概在5K左右,fastq文件18.3G,数据路径
https://www.10xgeznomics.com/datasets/5k-adult-mouse-heart-nuclei-isolated-with-chromium-nuclei-isolation-kit-3-1-standard
下载之后解压待用:
# 使用wget命令下载
wget -bc "https://cf.10xgenomics.com/samples/cell-exp/7.0.0/5k_mouse_heart_CNIK_3pv3/5k_mouse_heart_CNIK_3pv3_fastqs.tar"
# 解压下载的tar文件
tar -xvf 5k_mouse_heart_CNIK_3pv3_fastqs.tar分析流程:
首先是依据小鼠的参考基因组创建的比对索引文件"refdata-gex-GRCm39-2024-A",这个文件我们已经下载并进行了解压,它的数据结构如下所示:
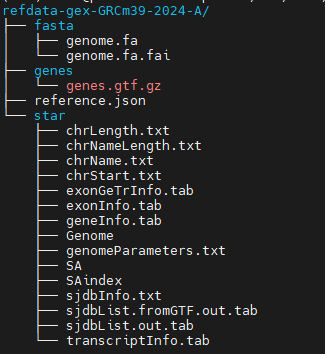
接着是我们在10X官网上下载的小鼠心脏的测试数据"5k_mouse_heart_CNIK_3pv3_fastqs",也已经进行了下载解压,数据都准备好之后就可以运行Cell Ranger了。
# 设置参考数据路径和测序文件路径
reference=/path/to/refdata-gex-GRCm39-2024-A
fq_path=/path/to/heart_10k_v3_fastqs
# 运行 Cell Ranger 计数分析
cellranger count --id=heart_10k_v3 # 样本名,需要根据自己样本信息更改
--transcriptome=${reference} # 索引文件路径,需要更改为对应物种的索引文件
--fastqs=${fq_path} # 测序文件路径,需要更改为自己的数据路径
--sample=heart_10k_v3 # 和样本名同名
--create-bam=true # 是否创建bam文件
--localcores=48 # 预设计算核心数,根据自己服务器的资源酌情设置
--localmem=150 # 预设内存占用,根据自己服务器的资源酌情设置
结果输出包含用于后续分析的矩阵文件filtered_feature_bc_matrix,对于其他的结果文件,我们不再一一解读。这是官网上的质控报告信息https://cf.10xgenomics.com/samples/cell-exp/7.0.0/5k_mouse_heart_CNIK_3pv3/5k_mouse_heart_CNIK_3pv3_web_summary.html)可以对比一下自己做的。以上就是通过Cell Ranger对下机数据的处理。
GEO平台数据
从GEO数据库(Gene Expression Omnibus)下载的数据通常有几种不同的格式,具体读取的方式取决于数据的存储形式。一般情况下,GEO的数据可以是原始数据(如 .CEL 文件)或处理后的数据(如 .txt、.csv、.tsv、.gz 格式的矩阵文件)。以下是读取GEO数据的几种常见方式,特别是如何将其导入Seurat进行分析。
1. 使用 GEOquery 包读取数据
GEOquery 是一个用于从GEO数据库下载和解析数据的R包,可以直接从GEO下载数据并将其导入到R中。这个包支持从GEO系列(GSE)、平台(GPL)以及样本(GSM)等信息中提取数据。
安装并加载 GEOquery 包
# 安装 BiocManager 包
install.packages("BiocManager")
# 使用 BiocManager 安装 GEOquery 包
BiocManager::install("GEOquery")
# 加载 GEOquery 包
library(GEOquery)下载并读取 GEO 数据集
假设你知道你想要下载的数据集的GSE号(比如GSE100000),你可以使用 getGEO() 函数从GEO数据库下载数据。
# 下载 GEO 数据
gse <- getGEO("GSE100000", GSEMatrix = TRUE) # GSEMatrix=TRUE表示获取矩阵数据
# 检查数据结构
str(gse)提取表达矩阵
从GEO数据集中,通常你会得到一个包含多个表达矩阵的列表。如果是多个系列数据,你可以选择感兴趣的一个。
# 提取第一个矩阵
expression_matrix <- exprs(gse[[1]])
# 如果有注释数据,你可以获取表型数据(样本信息)
pheno_data <- pData(gse[[1]])# 如果需要基因注释,可以提取基因的注释信息
feature_data <- fData(gse[[1]])BD(BD Biosciences)平台的数据
BD数据文件的格式
BD平台的数据一般是存储为以下几种格式:
.csv(Comma Separated Values):基因表达矩阵文件
.fcs(Flow Cytometry Standard):原始流式细胞术数据
.h5(HDF5):存储大规模数据的高效格式
.tsv(Tab-Separated Values):常见的分隔符格式
转换为Seurat输入格式
假设得到的是基因表达矩阵(通常是一个.csv或.tsv文件),可以按照以下步骤将其转换为Seurat需要的格式。
步骤 1: 读取数据
使用read.csv或read.table等函数读取矩阵数据。
# 读取基因表达矩阵
expression_data <- read.csv("path_to_your_data.csv", row.names = 1) # 假设基因在行,细胞在列
步骤 2: 读取条形码和基因信息
BD数据平台可能会提供条形码文件(通常是.csv或.tsv格式)和特征(基因)文件(也可能是.csv或.tsv格式)。你需要分别读取这些文件并将它们对应到表达矩阵中。
# 读取条形码(cells)信息
barcodes <- read.csv("path_to_barcodes.csv", header = FALSE)# 读取特征(基因)信息
features <- read.csv("path_to_features.csv", header = FALSE)步骤 3: 构建Seurat对象
Seurat需要一个稀疏矩阵来存储表达数据,同时需要条形码和特征信息。你可以使用CreateSeuratObject来构建Seurat对象。
# 加载 Seurat 包
library(Seurat)# 将基因表达矩阵转换为 Seurat 对象
seurat_obj <- CreateSeuratObject(counts = expression_data,project = "Your_Project_Name",assay = "RNA")这篇文章举了3个常用数据库平台的数据读取,如果大家遇到其他平台的数据读取问题,欢迎联系作者帮助解决!总的来说,大家获取数据的方式有两种,根据自己的研究目标、预算以及时间来决定是使用自己的测序数据还是依赖于公共数据库中的数据,无论哪种方式,弄懂上游分析对于下游分析有益无害哦~