【Scientific Data 】紫茎泽兰的染色体水平基因组组装
目录
写在前面
摘要
背景与概述
方法
植物材料与测序
单倍型解析的三倍体基因组从头组装
等位染色体间的结构变异
转座元件(TEs)注释
基因注释
数据记录
技术验证
写在前面
文章连接:Chromosome-level genome assembly of the crofton weed (Ageratina adenophora) | Scientific Data![]() https://www.nature.com/articles/s41597-025-04637-1本研究首次完成紫茎泽兰染色体水平基因组组装,解析关键变异与转座元件,揭示入侵遗传基础,为防控与研究提供重要资源。
https://www.nature.com/articles/s41597-025-04637-1本研究首次完成紫茎泽兰染色体水平基因组组装,解析关键变异与转座元件,揭示入侵遗传基础,为防控与研究提供重要资源。
摘要
紫茎泽兰(Ageratina adenophora)是一种重要的入侵物种,广泛破坏生态系统稳定性,造成重大经济损失。然而,由于缺乏基因组数据,目前对其入侵机制的遗传学认知仍然有限。在本研究中,我们利用PacBio Sequel长读长测序、光学图谱和Hi-C测序技术,成功完成了紫茎泽兰三倍体基因组的从头组装。最终组装得到一个包含51条染色体的单倍型解析基因组,总大小约为3.82 Gb,支架(scaffold)N50长度为70.8 Mb。BUSCO分析显示,97.71%的基因完整性得到确认。基因组注释结果表明,该基因组中含有3.16 Gb(占76.44%)的重复序列,并预测出123,134个蛋白质编码基因,其中99.03%已完成功能注释。这一高质量的参考基因组将为未来研究紫茎泽兰的进化动态及其入侵适应性提供宝贵的基因组资源。
背景与概述
生物入侵是全球普遍存在的问题,对生物多样性保护、生态稳定性和环境资源构成威胁,同时也带来严重的社会挑战与经济损失¹²。外来植物的迅速扩散,尤其是在国际贸易、旅游和交通运输快速发展的推动下,是生物入侵的重要组成部分,对社会科学、生态学和进化研究均具有重要意义。
紫茎泽兰(Ageratina adenophora(Spreng.) R.King & H.Rob.),俗称紫茎泽兰,是菊科(Asteraceae)的一种多年生常绿半灌,原产于中美洲。19世纪,它最初作为观赏植物被引入欧洲,随后扩散至澳大利亚和亚洲。如今,它已成为一种极具破坏性的全球性外来入侵杂草,在美国、澳大利亚、印度洋和太平洋岛屿,以及南亚和东亚地区广泛分布并造成严重问题。在中国,它被列为十大最具破坏性入侵物种之一。
紫茎泽兰具有多种有助于其成功入侵的生物学特性。首先,该植物含有多种活性物质,对其他植物具有强烈的化感作用,并对动物具有毒性,从而使其能在生态位中占据优势。例如,黄酮类化合物可作为抵御生物与非生物胁迫(包括草食动物、病原体、紫外线辐射和高温)的保护机制。此外,紫茎泽兰繁殖系数极高,每株植物可产生约10,000粒小而分散的种子。其对恶劣环境的高度适应能力、高效的土壤养分吸收能力,加上发达的根茎系统,使其能够快速入侵、定殖和扩张。
因此,全面理解这些生物学特性背后的遗传基础,不仅有望为高效防控紫茎泽兰提供科学依据,还能通过基因工程为作物改良提供有价值的基因资源。
尽管已有研究探讨了紫茎泽兰的生物学特性,但由于其遗传资源和基因组数据的匮乏,对其入侵机制的深入研究一直受到限制。在本研究中,我们采用多种测序技术和组装策略(图1),成功构建了紫茎泽兰的染色体水平参考基因组,其基因组大小约为3.82 Gb,支架N50为70.8 Mb。总之,本研究为进一步探索紫茎泽兰的入侵机制及制定有效防控策略提供了宝贵的基因组资源。
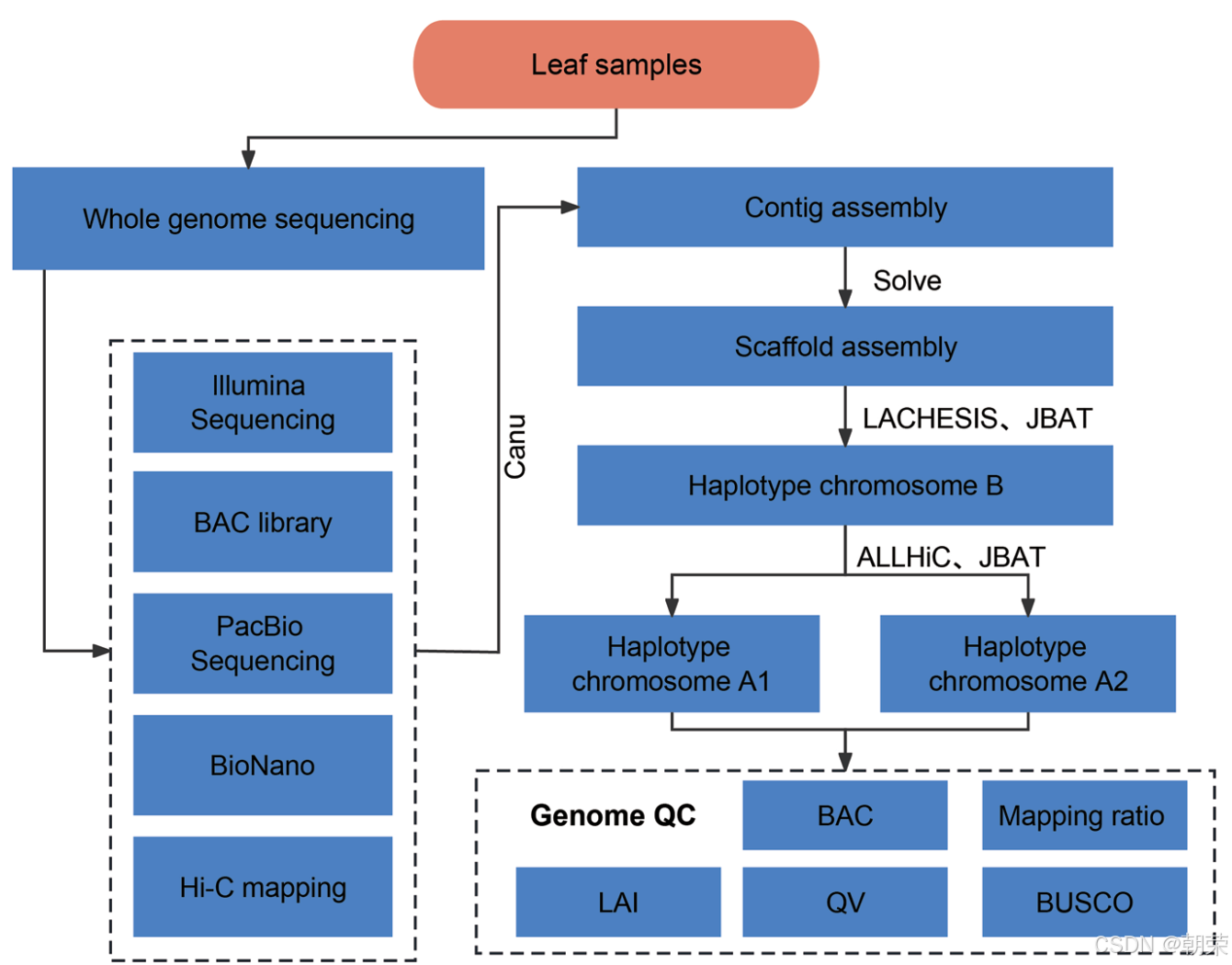
方法
植物材料与测序
本研究所用紫茎泽兰(Ageratina adenophora)样本采自中国云南省腾冲县(北纬25°52′9″204″,东经98°45′220″)。采样重点为幼嫩叶片,以获取高质量基因组DNA,用于后续基因组测序和Hi-C分析。紫茎泽兰基因组DNA的提取采用改良CTAB法⁹。DNA浓度与质量分别使用NanoDrop 2000(美国赛默飞世尔公司)和Qubit荧光计(美国赛默飞世尔公司)进行检测。
随后,按照标准Illumina建库流程,构建插入片段大小为270 bp的文库,并采用PE150测序策略在Illumina平台(美国)上进行测序,共获得92.83 Gb(约34×覆盖度)的高质量Illumina干净读段。按照PacBio推荐的建库方案(美国太平洋生物科学公司),构建插入片段大小为30 kb的PacBio DNA测序文库,并在PacBio Sequel平台上测序,获得原始数据总量为206 Gb(约54×覆盖度),平均读长为13,666 bp。同时,为进行基因组抛光分析,还生成了28.81 Gb的PacBio HiFi读段(平均读长为10,505 bp)。
BAC文库的构建与测序由南京弘远生物科技有限公司(中国南京)完成。Hi-C文库的构建参照先前文献方法¹⁰,并在Illumina平台上以2×150 bp读长进行测序,最终获得398.39 Gb(约104×覆盖度)的高质量Hi-C干净数据。采用BioNano Genomics Irys系统(美国BioNano Genomics公司)进行光学图谱构建,获得约797.39 Gb(约209×覆盖度)的高质量光学分子数据(分子长度>100 kb,标签信噪比≥3.0,平均分子强度<0.6)。
紫茎泽兰根、叶和花的总RNA提取与cDNA合成采用Yang等¹¹的方法。mRNA测序文库在Illumina NovaSeq平台上采用150 bp双端测序技术构建,每个样本设置三个生物学重复。全长转录组测序则按照PacBio Iso-Seq实验流程,对根、叶和花样本进行处理,并在PacBio Sequel平台上测序。
单倍型解析的三倍体基因组从头组装
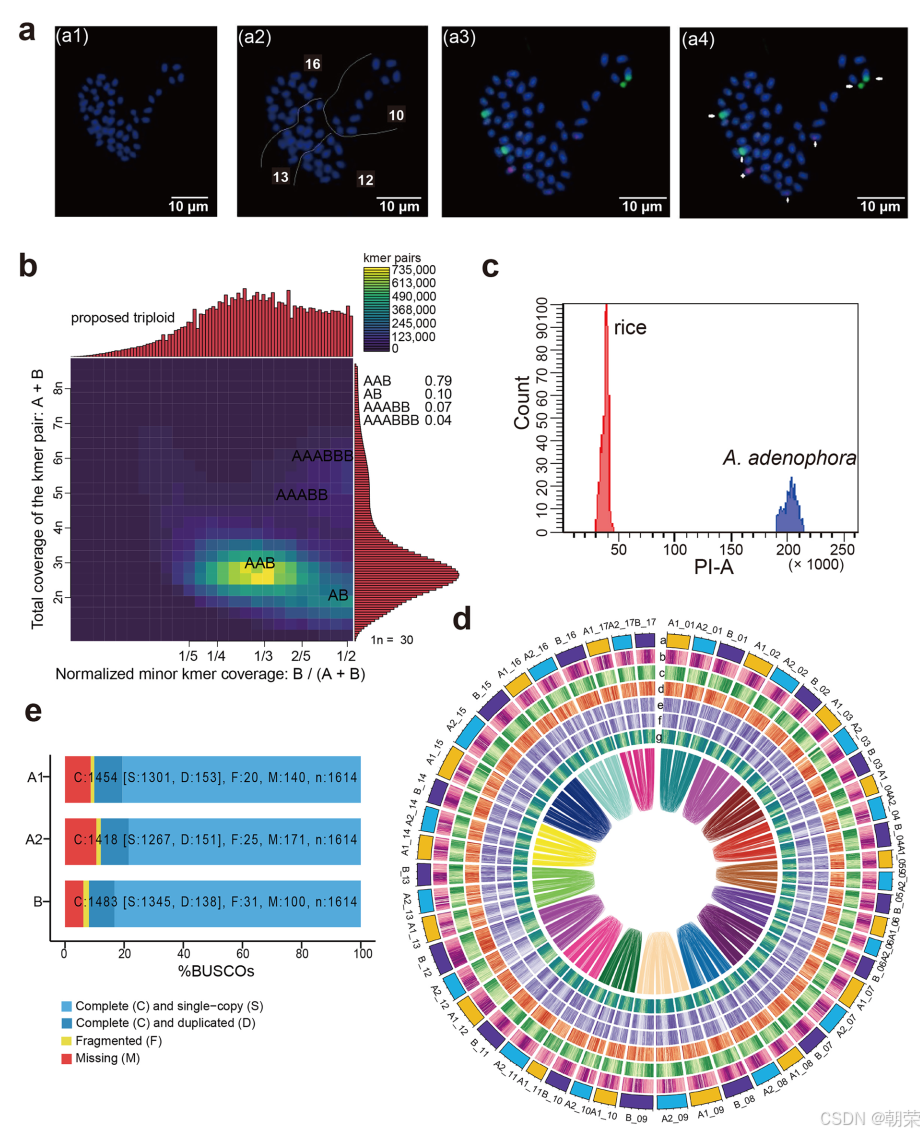
荧光原位杂交(FISH)技术确认紫茎泽兰具有51条染色体(图2a)。Smudgeplot¹²分析表明,紫茎泽兰具有“AAB”型基因组结构(图2b)。通过流式细胞术,并以水稻为标准参照基因组,估算紫茎泽兰的基因组大小。测得紫茎泽兰与水稻之间的荧光强度比值约为5.1,据此推算其基因组大小约为3.96 Gb(图2c)。
我们采用多种测序技术与精细的组装策略(图1),系统解析了紫茎泽兰基因组的复杂性。首先,利用Canu v1.9软件,基于PacBio Sequel的长读长数据进行Contig水平的组装(参数设置为genomeSize=3.96 g),并通过Pilon(v1.2229,参数:–min depth 10 –changes –fix bases)和Arrow v7.01(Pacific Biosciences)进行抛光,最终获得3.46 Gb的Contig序列,其N50为1.52 Mb。
随后,使用BioNano Solve v3.0.13进行杂交基因组组装(参数设置为“-B 2 -N 2”),超过93.35%的Contig序列被组装为超支架(super scaffolds),实现Scaffold N50为27.33 Mb。
接下来,我们使用LACHESIS¹⁵软件(版本20171221)进行染色体水平的初步组装,优化参数如下:CLUSTER_MIN_RE_SITES=100,CLUSTER_NONINFORMATIVE_RATIO=1.5,CLUSTER_MAX_LINK_DENSITY=2,ORDER_MIN_N_RES_IN_SHREDS=60,ORDER_MIN_N_RES_IN_TRUNK=60。通过对锚定染色体互作热图的分析,发现其中一个单倍型的互作信号明显强于另外两个。因此,我们利用Juicebox Assembly Tools(JBAT v1.1)¹⁶软件,从三个嵌合单倍型中分离出互作信号更强的单倍型,最终获得一个非冗余的、单倍型解析的组装结果,包含17条染色体,命名为单倍型B。
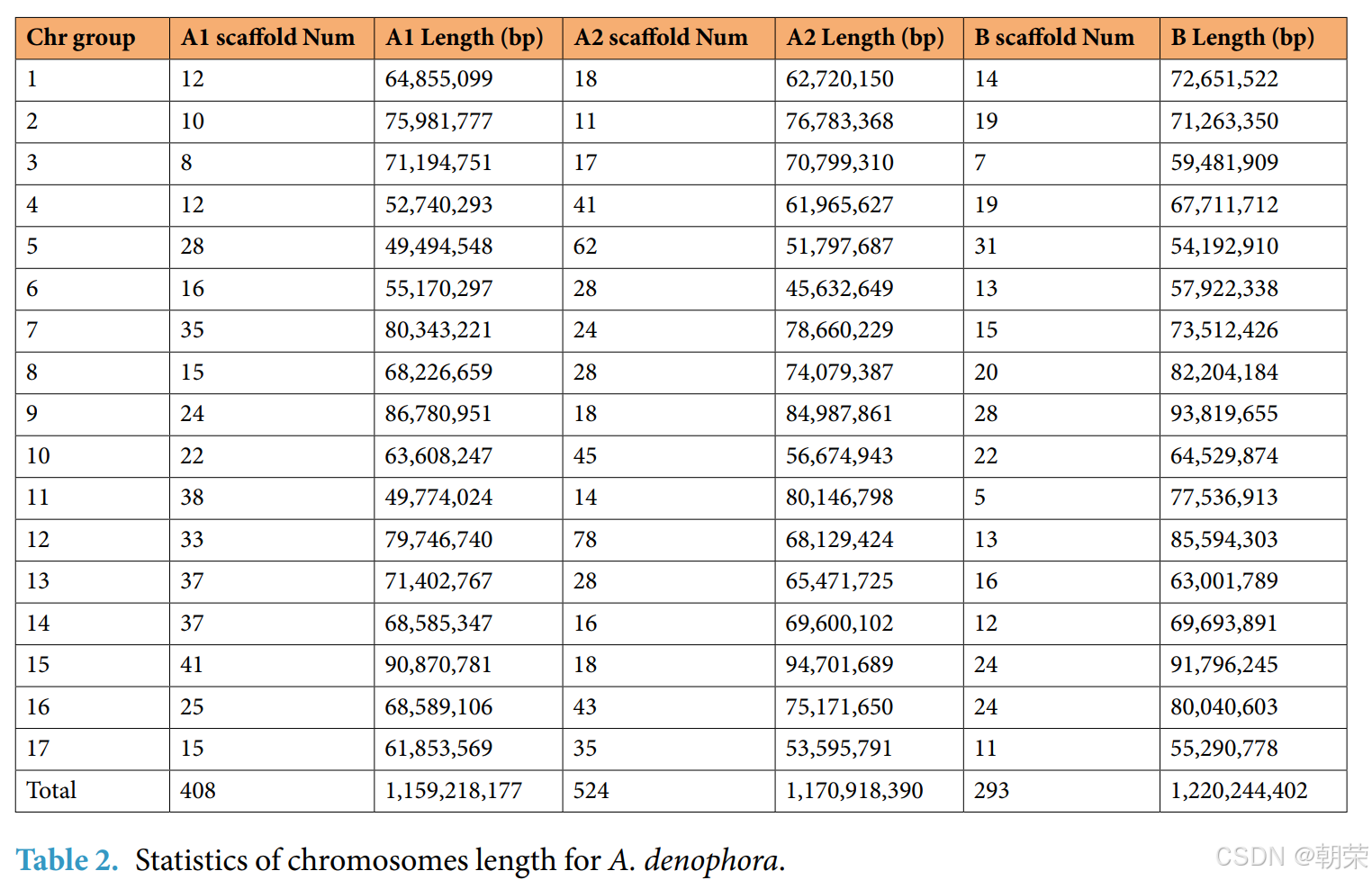
以单倍型B为参考,我们进一步采用ALLHIC¹⁷方法区分其余两个单倍型A1和A2。最终,我们得到了紫茎泽兰的单倍型解析、染色体水平的基因组组装,总大小约为3.82 Gb,Scaffold N50为70.8 Mb(图2d与表1)。值得注意的是,该组装结果覆盖了流式细胞术估算基因组大小的96.46%(图2c)。其中,高达3.55 Gb(占总组装的92.98%)的Scaffold成功锚定到51条伪染色体上。这些伪染色体被划分为17个同源群,每个同源群包含三条等位染色体(A1:1.16 Gb,A2:1.17 Gb,B:1.22 Gb)(表2)。
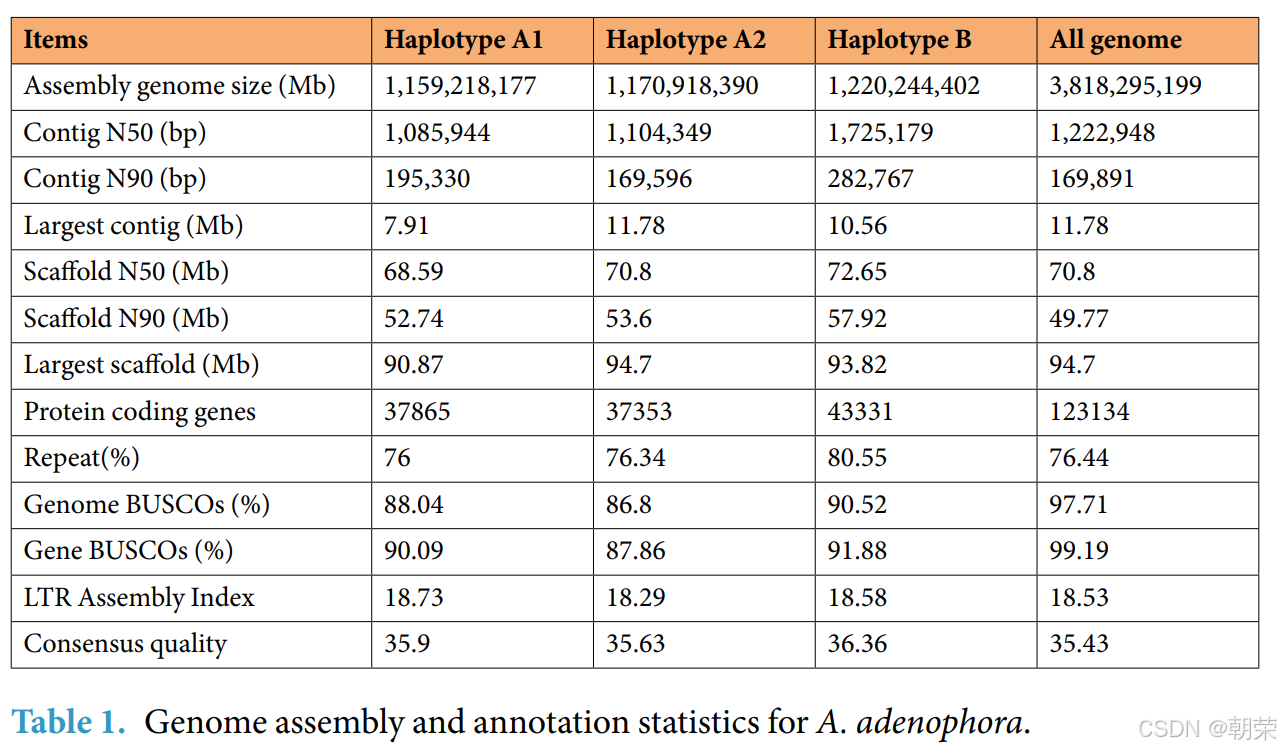
基准通用单拷贝直系同源基因(BUSCO)¹⁸分析显示,本组装中约97.71%的BUSCO基因完整(图2e)。LTR结构分析显示,该基因组的LTR组装指数(LAI)为18.53(表1)。利用Merqury²⁰软件包(v1.3),基于紫茎泽兰的短读长测序数据对基因组质量和完整性进行评估,结果显示基因组碱基准确率超过99.97%(QV > 35.43),k-mer完整性为97.30%(表1)。
综上,这些结果充分表明,我们构建的紫茎泽兰基因组组装具有很高的质量和完整性。
等位染色体间的结构变异
我们采用 MUMMER v4.021 软件对不同单倍型之间的结构变异进行分析。首先使用 “nucmer” 命令进行基因组比对,参数设置为 “–maxmatch -c 500 -b 500 -l 100”。随后,应用 delta-filter 命令对比对结果进行优化,参数设置为 “-i 90 -l 1000 -m”,以确保最小匹配长度为 1 Kb 且相似度不低于 90%。接着,使用 SyRI(v1.6)22 软件识别三个单倍型之间的结构变异,参数设置为 “–allow-offset 100 –unic 2000”。为确保结果准确性,仅保留与 SYN(同源区域)重叠比例不低于 50% 的 DUP(重复)类型变异,且所有变异的最小长度要求为 30 Kb。
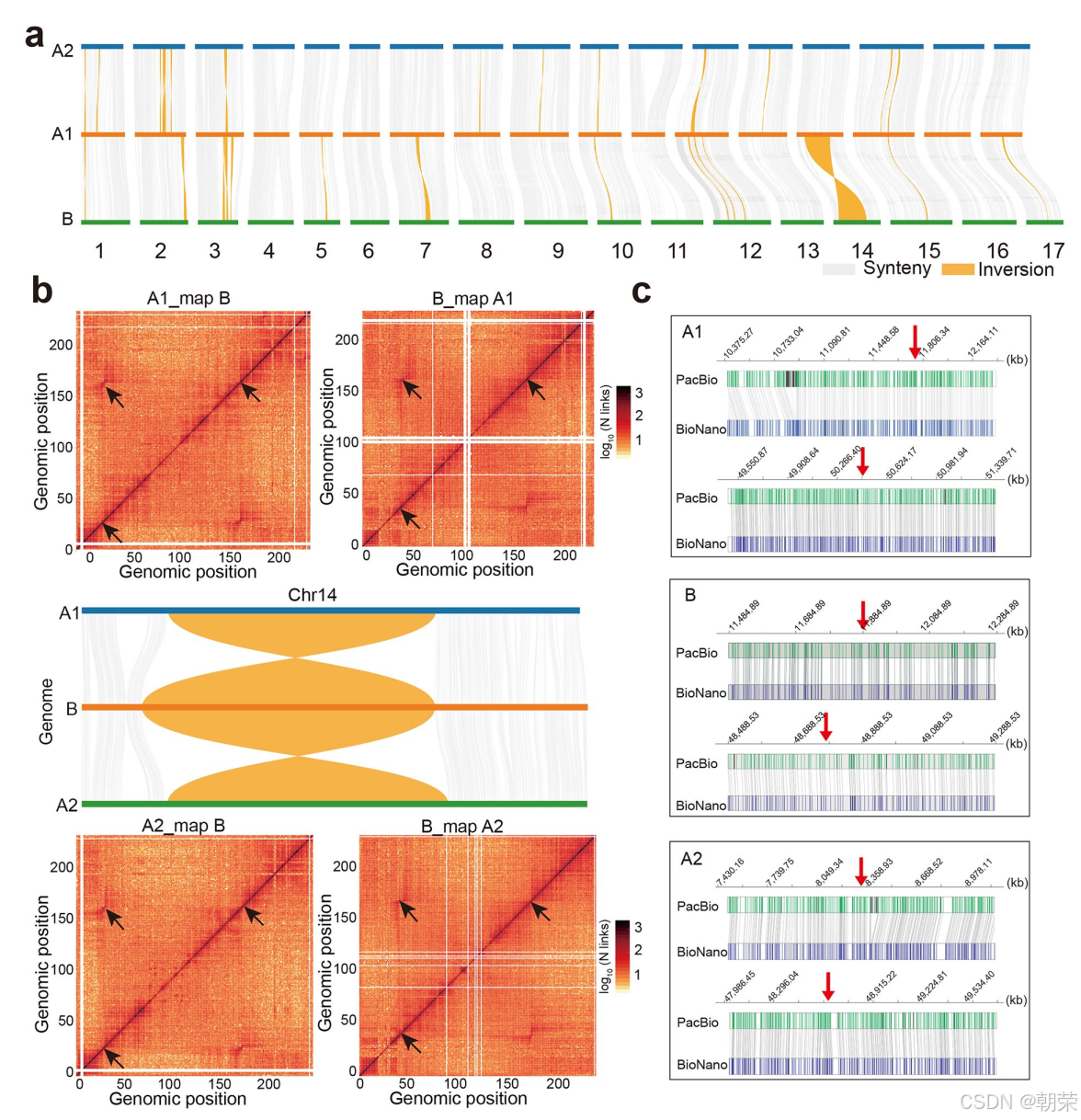
最终,共检测到 1,352 个长度大于 30 Kb 的单倍型间结构变异。值得注意的是,还发现了 46 个超大型倒位变异(>1 Mb),这些倒位变异累计占所有结构变异总长度的 约 57.80%。其中,最大的倒位变异发生在第 14 号染色体上,跨度约 40 Mb,占该染色体总长度的 57.88%(图 3a)。Hi-C 和光学图谱技术进一步验证了这些倒位变异的准确性(图 3b 和图 3c)。
转座元件(TEs)注释
为构建紫茎泽兰(Ageratina adenophora)全面的重复序列库,我们使用 RepeatModeler23(v2.02)、LTR-FINDER24(v1.05)、MITE-hunter(20100819)25 和 PILER-DF26(v1.0)等工具,并采用默认参数进行分析。随后将该序列库与 Repbase27 数据库进行合并,并利用 PASTEClassifier.py28 软件将序列分类至不同类别。最后,使用 RepeatMasker v4.1.129 软件,基于最终构建的重复序列库对基因组进行遮蔽(masking)处理。
最终,在紫茎泽兰基因组中共鉴定出 3.16 Gb(占基因组总长的 76.44%)的重复序列(表 3)。其中,单倍型 A1、A2 和 B 中的转座元件(TEs)含量分别为 881.04 Mb(76.00%)、893.83 Mb(76.34%)和 982.94 Mb(80.55%)。
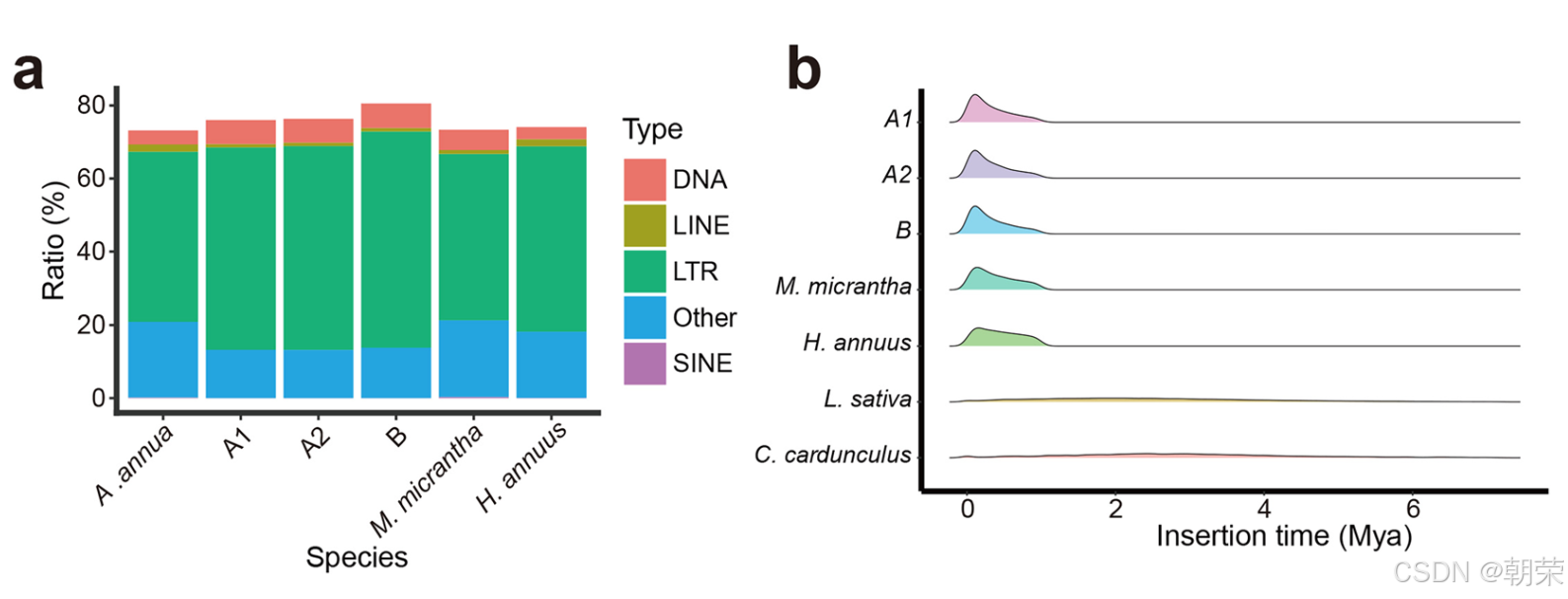
通过与几种近缘菊科(Asteraceae)物种的比较分析,我们发现紫茎泽兰中 LTR 类转座元件的比例显著偏高(图 4a)。为进一步阐明 LTR 的扩张机制,我们利用 LTRretriever30(v2.9.8)在紫茎泽兰中鉴定了 55,232 个全长 LTR-逆转录转座子(LTR-RTs),其中单倍型 A1、A2 和 B 分别包含 17,721、17,646 和 19,865 个。这些 LTR-RTs 中,61.71% 属于 Gypsy 亚家族,16.89% 属于 Copia 亚家族。
此外,我们还计算了 LTR 的插入时间。通过使用 MAFFT31(v7.205,参数:–local pair –max iterate 1000)对 LTR 两侧的旁侧序列进行比对,随后利用 EMBOSS32(v6.6.0)中的 Kimura 模型计算序列距离(K)。LTR 插入时间的计算公式为:T = K / (2 × r),其中分子钟速率(r)设为 7 × 10⁻⁹。分析结果显示,三个单倍型均经历了一次近期爆发式扩张(< 0.5 百万年前,即 <0.5 Mya),该扩张事件与向日葵(Helianthus annuus)和微型蔓长春花(Micranthemum micranthemoides)的情况一致,但晚于莴苣(Lactuca sativa,约 1.9 Mya)和菜蓟(Cynara cardunculus,约 2.4 Mya)(图 4b)。
基因注释
我们综合运用了同源比对法、从头预测法以及转录组预测法对紫茎泽兰(Ageratina adenophora)基因进行注释。其中,从头预测使用了 GenScan33(v1.0)、Augustus34(v2.4)、GlimmerHMM35(v3.0.4)、GeneID36(v1.4)和 SNAP37 等软件;同源比对预测则采用 GeMoMa38(v1.4.2),并参考了日本栽培稻(Oryza sativa L. ssp. japonica)、拟南芥(Arabidopsis thaliana)、小花蔓泽兰(M. micrantha)和向日葵(H. annuus)的基因组信息。
此外,我们使用 HISAT39(v2.1.0)将来自不同组织的干净 RNA-seq 读段比对到基因组上,随后将这些比对结果输入 Trinity40(v2.2.0)进行基因组引导模式下的从头转录组组装,生成 unigenes(转录本聚类单元)。这些 unigenes 与 PacBio 全长 cDNA 一起,再通过 BLAT41(v35)比对回基因组,并利用 PASA v2.0.242 进行转录本预测。
最后,使用 EVidenceModeler v1.1.143 将同源比对、转录组和从头预测的结果进行整合,生成统一的基因模型,并通过 PASA 进行更新。所有上述软件工具的最大内含子长度均设置为 20 千碱基(kb)。最终,我们在紫茎泽兰基因组中预测并鉴定出 123,134 个蛋白质编码基因(表 1)。
在基因功能注释方面,我们使用 DIAMOND v0.9.2844 将预测的蛋白质序列与 NCBI 非冗余蛋白质数据库(NR)、eggNOG45、SwissProt 和 TrEMBL46 数据库进行比对,比对阈值设为 1e-5。利用 HMMER47(v3.1b2)搜索 Pfam48 数据库进行蛋白质结构域注释,基因本体(Gene Ontology, GO)注释则通过 InterProScan50(v4.3)获取。此外,京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes, KEGG)通路注释通过 KEGG 自动注释服务器(KAAS)5152 完成。最终,约有 99.03%(121,934 个)的基因模型在 SwissProt、TrEMBL、NR、KEGG、GO、eggNOG 或 Pfam 数据库中得到了功能注释(表 4)。
数据记录
本研究中所使用的所有原始测序数据⁵³以及全基因组鸟枪法(Whole Genome Shotgun, WGS)组装结果⁵⁴均已通过 BioProject 编号 PRJNA1096832 提交至美国国家生物技术信息中心(NCBI)。其中:
- Illumina 基因组测序数据存储于 NCBI 序列读取档案库(Sequence Read Archive, SRA),编号为 SRR28607150 和 SRR28607151;
- PacBio CLR(连续长读长)DNA 测序数据的 SRA 编号为 SRR28607117;
- PacBio HiFi DNA 测序数据的 SRA 编号为 SRR28607109 和 SRR28607110;
- PacBio Iso-Seq(全长转录组)数据(涵盖花、根和叶组织)的 SRA 编号分别为 SRR28607137、SRR28607138 以及 SRR28607140–SRR28607143;
- RNA-seq 数据(花、根和叶组织)的 SRA 编号为 SRR28607106–SRR28607108 以及 SRR28607144–SRR28607149;
- Hi-C 测序的短读长数据的 SRA 编号为 SRR28607128 和 SRR28607139。
最终的基因组组装结果、结构变异、转座元件、基因结构与功能注释数据已存入 Figshare 数据库。
技术验证
我们对基因组的连续性进行了评估,结果显示contig N50 值达到了 1.22 Mb(表 1),相比之前报道的三倍体香蕉基因组(contig N50 为 1.08 Mb)⁵⁶有显著提升。Benchmarking Universal Single-Copy Orthologs(BUSCO)分析显示,本组装中约 97.71% 的 BUSCO 基因是完整的(表 1 和图 2e),与栽培六倍体菊花(C. morifolium)⁵⁷的报道比例相当。长末端重复序列(LTRs)是评估重复序列及基因间区组装质量的重要指标。LTR 注释分析显示,紫茎泽兰基因组的 LTR 组装指数(LAI)为 18.53(表 1),达到了参考基因组的标准要求⁵⁸。通过与紫茎泽兰全基因组短读长测序数据的比对,我们确认该基因组的碱基准确率超过 99.97%(QV > 35.43),k-mer 完整性为 97.30%(表 1)。
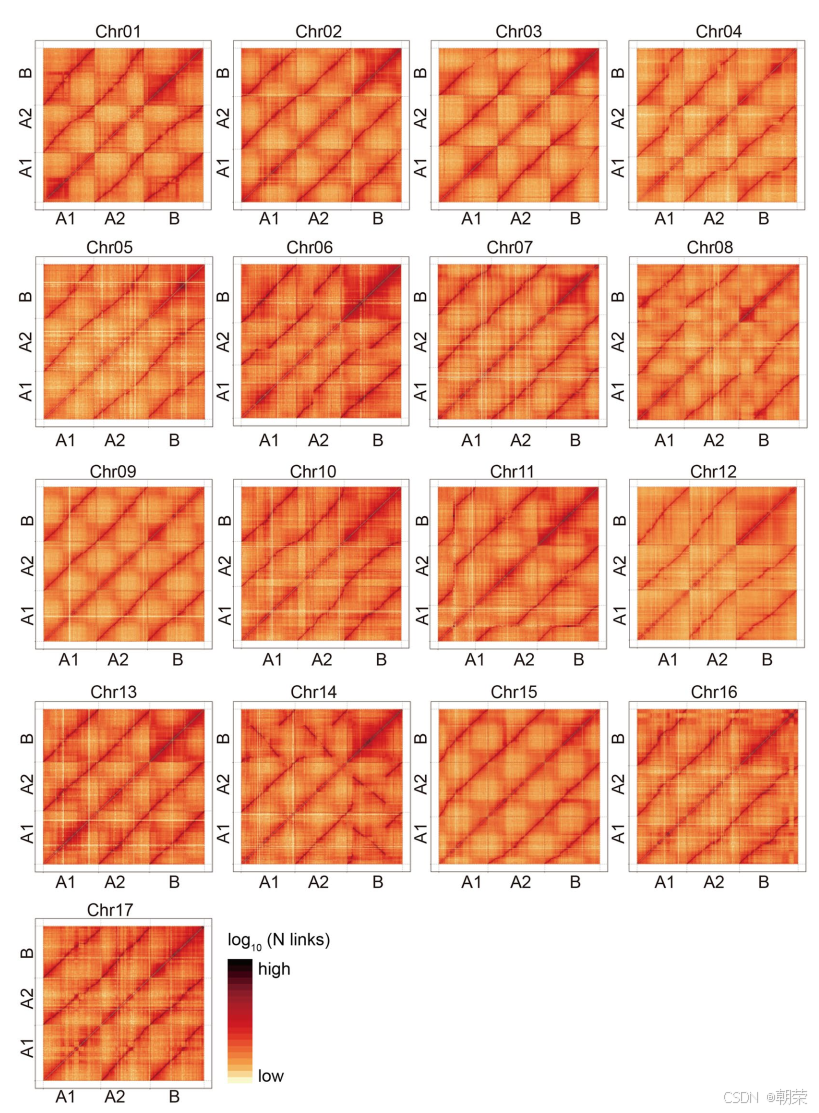
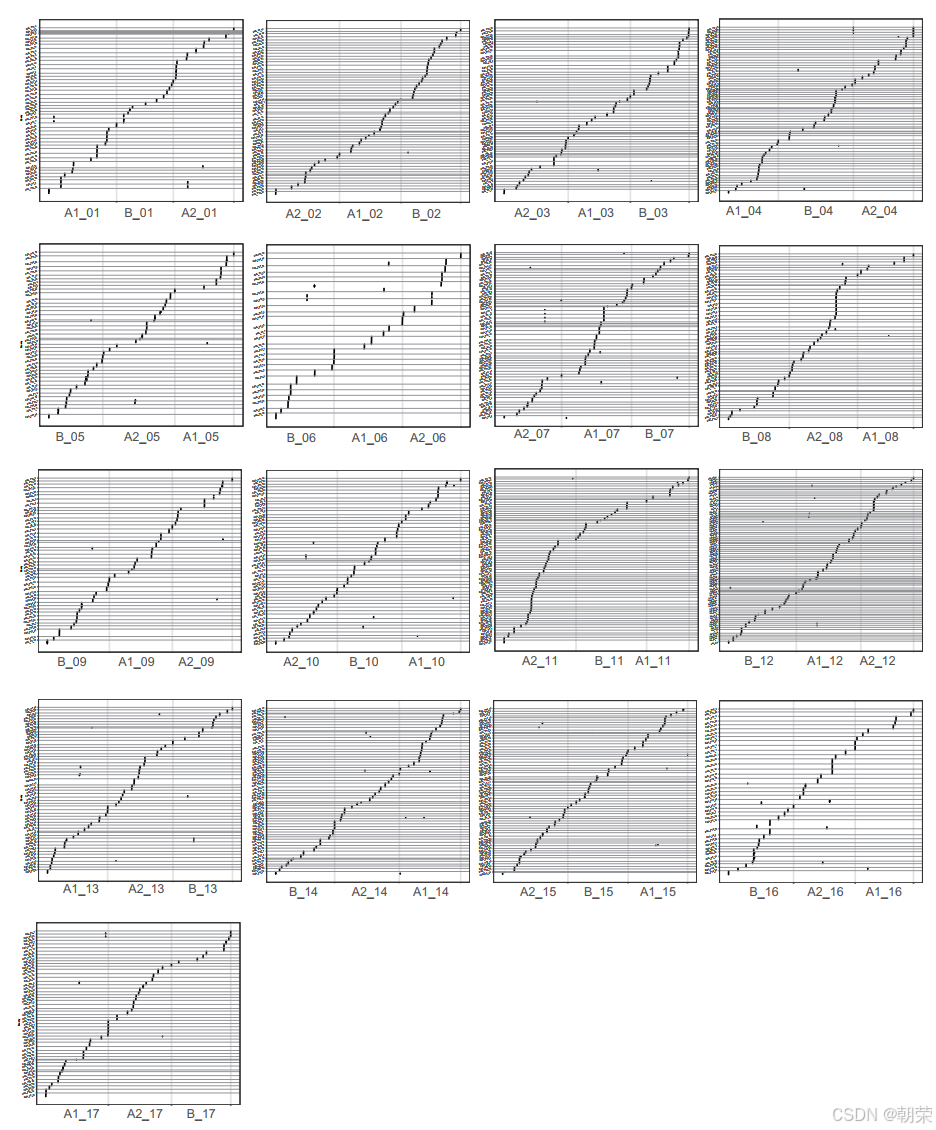
染色体互作热图显示,51 条伪染色体被归为 17 个同源群(homoeologous clusters),每个群包含三条等位染色体(图 5)。为进一步验证单倍型解析基因组的准确性,我们利用 971 条细菌人工染色体(BAC)序列(N50 为 114.26 kb),计算出单倍型间切换错误率(switch error)为 4.20%(图 6)。
其中,单倍型 B 与 A 之间最大的倒位变异位于第 14 号染色体,跨度约 40 Mb,占该染色体总长度的 57.88%(图 3a)。通过单倍型间 Hi-C 数据的映射,我们在断裂点周围观察到了离散的染色质互作信号(图 3b),并且这些倒位变异也通过 BioNano 光学图谱得到了验证(图 3c)。