【数据分析】微生物组数据的批次校正与分析
禁止商业或二改转载,仅供自学使用,侵权必究,如需截取部分内容请后台联系作者!
文章目录
-
- 介绍
-
- 数据准备
- ConQuR方法的应用
- 校正效果的评估
- 预测能力的评估
- 参数调优
- 加载R包
- 导入数据
- 数据预处理
- 默认运行 ConQuR
- 施加惩罚向运行 ConQuR
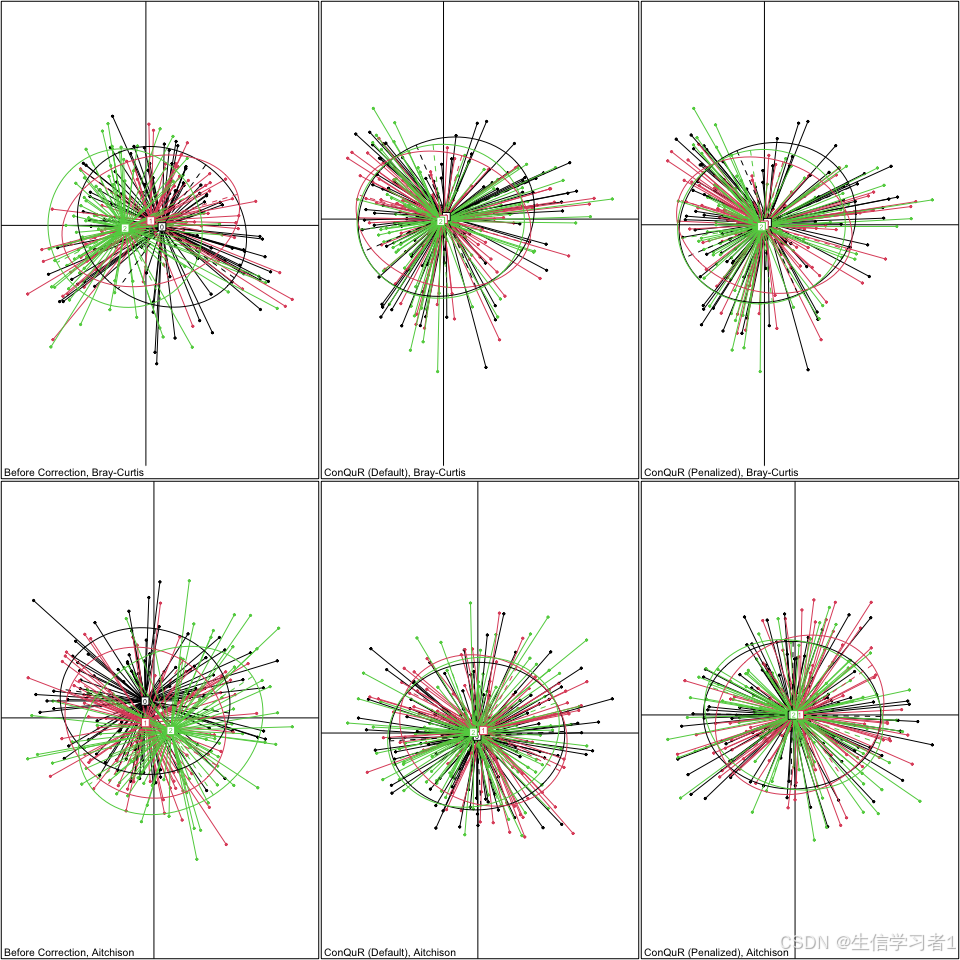
- 评估效果
- 调节参数
- 总结
- 系统信息
介绍
在微生物组学研究中,由于实验条件、测序平台或样本处理等差异,数据中常常存在批次效应。这些批次效应可能会掩盖真实的生物学信号,从而影响研究结果的准确性和可靠性。因此,对微生物组数据进行批次校正至关重要。本文介绍了一种基于R语言的微生物组数据批次校正方法——ConQuR(Compositional Quantile Regression),并通过一系列分析流程展示了其在实际数据中的应用效果。
ConQuR 软件包(版本 2.0)包含一系列函数,可利用条件分位数回归方法从属种读取计数表中去除批次效应。该软件同时考虑了微生物组数据的分布特征——零膨胀和过度离散。为了最大程度地去除批次效应,提供了在 ConQuR 变化范围内的参数调整功能。此外,还提供了用于计算 PERMANOVA R2、打印 PCoA 图以及基于属读取计数表预测关键变量的支持函数。
以下这些包是 ZINQ 包中相关功能和示例所必需的:quantreg、cqrReg、glmnet、dplyr、doParallel、gplots、vegan、ade4、compositions、randomForest、ROCR、ape、GUniFrac、fastDummies。
数据准备
首先,我们使用了S