gro文件和top文件介绍,以及如何合并两个gro文件或两个top文件
关于.gro文件和.top文件
- gro文件:包含分子系统的原子坐标、速度等结构信息,是GROMACS中的坐标文件,类似于PDB文件,但格式不同。
- .top文件:包含分子系统的拓扑信息,定义了分子中的原子类型、电荷、键合参数等力场信息。
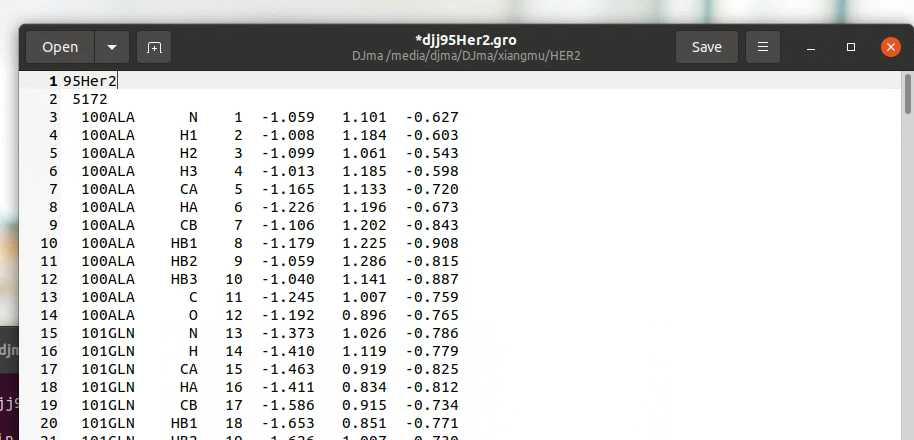
gro文件格式通常包含:
- 第一行:标题或注释
- 第二行:原子总数
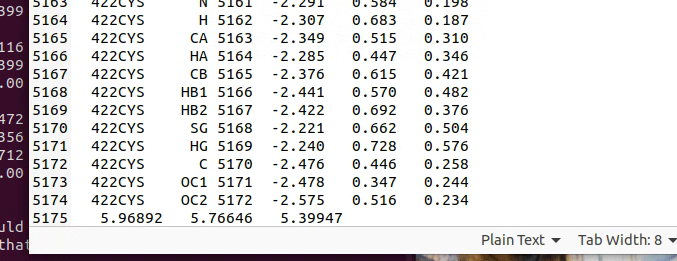
- 中间部分:每行包含残基编号、残基名称、原子名称、原子编号、xyz坐标(单位nm)以及可选的速度
- 最后一行:盒子尺寸信息
合并两个分子的gro文件
- 将两个.gro文件中的坐标部分(除去头尾信息)合并到一个文件中
- 更新原子总数
- 确保盒子尺寸合适(3.1避免多个分子(蛋白)结构之间重叠;3.2离盒子边界有一定距离)
特殊案例:(如果是多个分子或者多个蛋白的体系)
# 准备第一个蛋白质结构和盒子
gmx editconf -f protein1.gro -o protein1_box.gro -c -d 2.0 -bt dodecahedron
# 调整第二个蛋白质的位置(如果需要特定相对位置)
gmx editconf -f protein2.gro -o protein2_positioned.gro -translate 0 0 6 # 在z轴方向移动6nm
# 合并两个蛋白质
gmx insert-molecules -f protein1_box.gro -ci protein2_positioned.gro -o complex.gro -try 100
# 根据合并后的蛋白质复合物大小重新定义盒子
gmx editconf -f complex.gro -o complex_final.gro -c -d 2.0 -bt dodecahedron