【每天一个知识点】单细胞RNA-seq数据注释综述
一、研究背景与意义
单细胞RNA测序(single-cell RNA sequencing, scRNA-seq)作为高通量测序技术的重大突破,能够在单细胞水平捕获转录组信息,刻画细胞间的异质性,推动了发育生物学、免疫学、肿瘤学等领域的革命性进展。与传统的群体测序相比,scRNA-seq不仅揭示了组织中存在的细胞类型组成,还能解析细胞亚群、发育轨迹以及疾病状态下的转录特征。
然而,scRNA-seq实验产生的数据量庞大,通常表现为细胞 × 基因的稀疏表达矩阵。如何从这些高维且噪声显著的数据中,准确地将细胞归类为已知的 细胞类型(cell type) 或 细胞状态(cell state),是数据分析流程中的核心环节。这个过程被称为 单细胞RNA-seq数据注释(annotation)。
注释不仅是数据解读的必要条件,也是后续生物学发现的基础。例如:
在免疫系统研究中,准确区分 T 细胞亚群对于理解免疫反应和免疫治疗至关重要;
在癌症研究中,识别肿瘤微环境中的免疫抑制细胞群体有助于揭示耐药机制;
在干细胞与发育研究中,注释不同发育阶段的细胞群能为组织再生和疾病建模提供依据。
因此,开发和优化高效、准确、通用的单细胞注释方法,已成为当前生物信息学与计算生物学的重要研究方向。
二、常见的注释方法
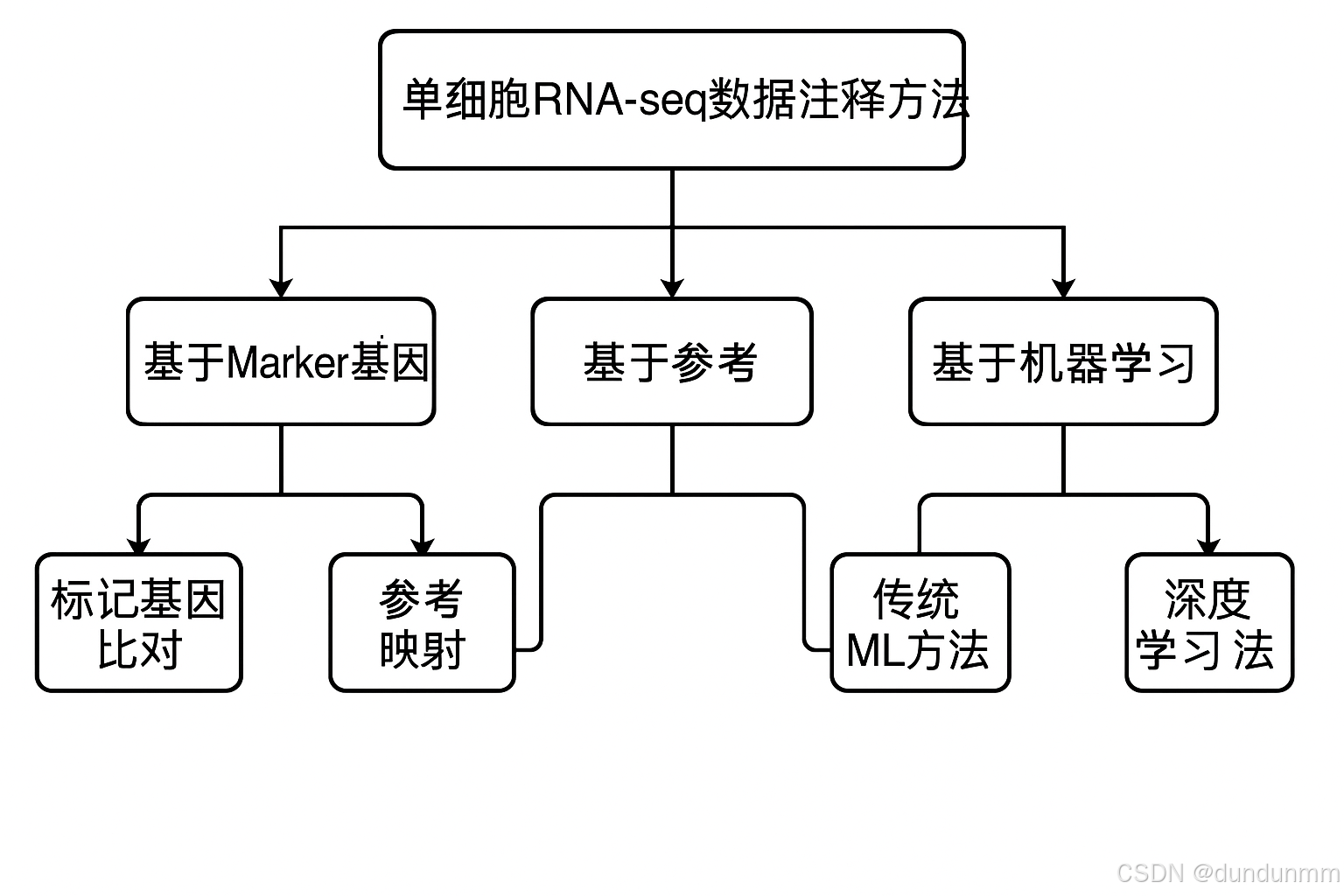
目前,单细胞RNA-seq数据注释的方法主要分为以下几类:
1. 基于标记基因(Marker Gene-based Annotation)
这是最传统、直观的方法。研究者根据已有的生物学知识或公开数据库(如 CellMarker、PanglaoDB),提取具有细胞特异性的标记基因。例如:
CD3E、CD4、CD8A 用于区分不同T细胞亚群;
PECAM1、VWF 标记内皮细胞;
GFAP 标记星形胶质细胞。
优点:简单直观,易于解释。
缺点:依赖人工经验,难以扩展到大规模数据;对稀有细胞或未知细胞类型不适用。
2. 基于聚类与差异分析(Clustering + DEGs)
利用无监督聚类(如 k-means、Louvain、Leiden)将细胞划分为群集,然后通过差异表达分析(DEGs)提取特征基因,并与已知标记库比对,推断细胞类型。
优点:能够发现潜在的新亚群。
缺点:聚类结果依赖参数和算法,对数据质量敏感;仍需人工解读。
3. 基于参考图谱的比对方法(Reference Mapping)
随着 Human Cell Atlas 等大规模参考数据集的构建,越来越多的方法采用“参考-查询”模式,将新数据与参考图谱进行匹配:
SingleR:基于相关性计算,选择与参考样本最相似的细胞类型。
scmap:基于最近邻搜索,将单细胞投射到参考数据。
Seurat Transfer Anchors:利用锚点(anchors)实现跨数据集映射。
优点:能够利用已有知识库,减少人工工作量。
缺点:依赖参考图谱的质量;当参考集覆盖不足时,准确性下降。
4. 基于机器学习和深度学习的方法
近年来,人工智能技术逐渐被引入单细胞注释,提升了自动化与泛化能力。
监督学习方法:
ACTINN:基于神经网络的细胞分类器。
scPred:利用支持向量机(SVM)进行预测。
深度学习方法:
scDeepSort:结合图神经网络(GNN)实现细胞类型注释。
Cell BLAST:通过深度表示学习实现相似性搜索。
SELINA:采用多对抗域自适应网络(MDA),解决批次效应并增强稀有细胞类型的注释。
优点:自动化程度高,能够处理大规模数据,具有较强的适应性。
缺点:模型训练复杂,需要较大的计算资源;可解释性较低。
三、面临的主要挑战
尽管方法不断进步,但单细胞RNA-seq数据注释仍存在若干难题:
1. 批次效应(Batch Effect)
不同实验条件、测序平台或样本来源会引入系统性差异,导致相同类型的细胞在不同数据集中表现出较大差异。如何有效消除批次效应,是提高跨数据集注释准确性的关键。
2. 注释标准不统一
细胞命名体系在不同研究之间往往缺乏一致性,例如同一类细胞在不同数据库中可能被标记为不同名称,造成信息整合的困难。
3. 稀有细胞与新细胞类型识别
稀有细胞类型在数据中占比极低,容易被聚类算法忽略或被误判。同时,新发现的细胞类型缺乏标记基因和参考图谱,增加了注释难度。
4. 疾病背景的复杂性
疾病状态下的细胞往往具有显著不同的转录特征,传统的参考数据大多来自健康组织,难以直接应用于疾病样本注释。
5. 可解释性与可复现性
深度学习模型虽能提供高准确率,但其“黑箱”特性限制了生物学解释。此外,注释流程的标准化程度不足,影响结果的复现性和可比性。
四、发展趋势与未来方向
1. 构建统一的参考图谱
未来的发展将依赖于大规模、多组织、多状态的标准化参考数据库。例如 Human Cell Atlas、Tabula Sapiens 等项目正不断扩展,推动形成统一的注释体系。
2. 多模态与跨组学融合
单细胞组学正在向多模态发展,如 scATAC-seq、空间转录组学(spatial transcriptomics)、蛋白质组学。融合不同模态数据(RNA+ATAC+空间位置信息)将为细胞注释提供更全面的证据。
3. 引入生成模型与数据增强
针对稀有细胞类型,利用生成对抗网络(GAN)、变分自编码器(VAE)进行数据增强与模拟,有助于缓解类别不平衡问题。
4. 注释框架自动化与可解释化
未来的方法将趋向于端到端的自动化流程,同时加强可解释性,结合标记基因知识和模型输出,提升生物学信任度。
5. 疾病背景下的特异性注释
开发面向癌症、免疫疾病、神经退行性疾病等特定场景的专业注释模型,能够提升在临床研究中的应用价值。
五、总结
单细胞RNA-seq数据注释是连接原始测序数据与生物学发现的关键步骤。当前方法涵盖了标记基因比对、无监督聚类、参考图谱映射以及深度学习模型等多种路径。尽管在自动化、准确性和大规模应用方面取得了显著进展,但批次效应、稀有细胞识别、疾病状态复杂性等问题仍然存在。
未来的发展趋势将集中在 统一参考图谱、多模态数据融合、生成模型应用、自动化与可解释化框架 等方面。随着数据量的持续积累和算法的不断优化,单细胞注释有望迈向更加全面、标准化与智能化的阶段,从而为精准医学和基础生物学研究提供强有力的支撑。