GEARS以及与基础模型结合
理解基因扰动的反应是众多生物医学应用的核心。然而,可能的多基因扰动组合数量呈指数级增长,严重限制了实验探究的范围。在此,图增强基因激活与抑制模拟器(GEARS),将深度学习与基因-基因关系知识图谱相结合,利用扰动筛选的单细胞RNA测序数据,预测单细胞对单基因和多基因扰动的转录反应。GEARS能够预测从未经实验扰动的基因组合的扰动结果。GEARS可以预测多基因扰动的表型差异效应,从而指导扰动实验的设计。
Predicting transcriptional outcomes of novel multigene perturbations with GEARS,Nature Biotechnology,2023
目录
- 背景概述
- GEARS框架
- 预测新的生物学表型
- scFoundation+GEARS
- 补充内容:化学扰动和基因扰动(参考UniPERT)
背景概述
细胞对基因扰动的转录反应揭示了细胞功能的基本机制。转录反应可描述多种功能,从基因调控机制如何维持细胞特性到调节基因表达如何逆转疾病表型。这对生物医学研究具有重要意义,尤其是在开发个性化疗法方面。例如,通过基因扰动研究验证药物靶点可提高临床试验成功的可能性。此外,识别协同基因对可增强联合治疗的效果。已知复杂的细胞表型由少数基因之间的遗传相互作用产生,因此识别此类相互作用可促进精准细胞工程的发展。尽管近年来的技术进步使科学家能够更快速地通过实验获取扰动结果,但由于潜在多基因组合的数量呈指数级增长,预测扰动效应的计算方法对于缩小实验规模至关重要。
然而,现有的预测扰动结果的计算方法自身存在局限性。预测单基因扰动结果的主流方法依赖于以基因调控网络的形式推断基因之间的转录关系。这种方法的局限性在于,要么难以从基因表达数据集中准确推断网络,要么从公共数据库中获取的网络不完整。此外,使用此类网络构建的现有预测模型通过线性组合单个扰动的效应进行预测,这使得它们无法预测多基因扰动的非加性效应(如协同作用)。最近的研究利用在大规模扰动筛选数据上训练的深度神经网络,跳过网络推断步骤,将遗传关系直接映射到潜在空间以预测扰动结果(比如scGen)。然而,这些方法仍要求组合中的每个基因在预测组合扰动效应之前必须经过实验扰动(来自已知细胞类型的对照和刺激数据)。
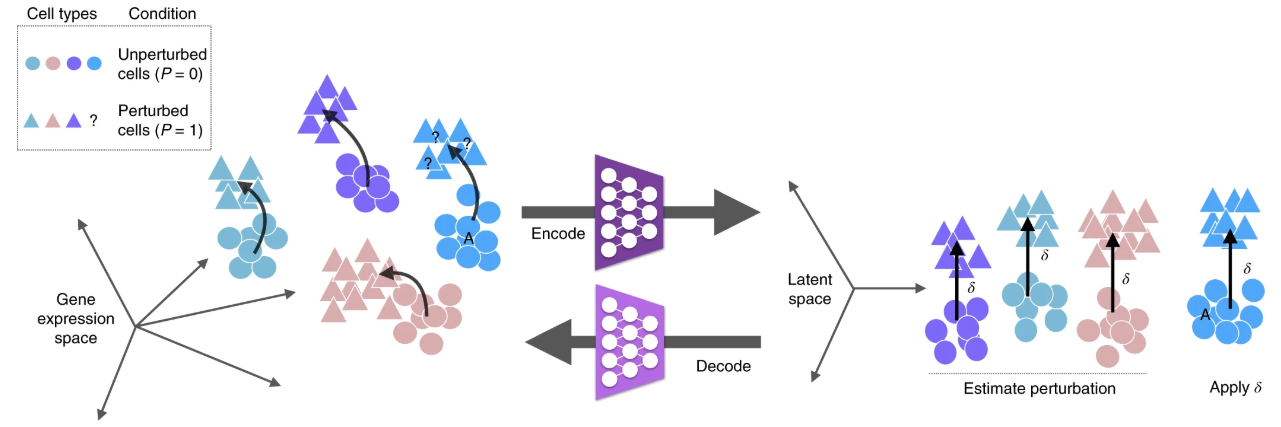
- scGen:在给定对照组和刺激组中一组已观察到的细胞类型的情况下,我们旨在通过训练一个能够学习训练集中细胞响应的模型,来预测新细胞类型A(蓝色)的扰动反应。在scGen模型中,该模型为变分自动编码器(VAE),其预测通过在自动编码器的潜在空间中进行向量运算获得。具体而言,我们使用编码器网络将基因表达测量值投影到潜在空间,并获得向量δ,该向量表示训练集中受扰动细胞与未受扰动细胞在潜在空间中的差异。利用向量δ,对A类型的未受扰动细胞在潜在空间中进行线性外推,然后通过解码器网络将潜在空间中的线性预测映射到基因表达空间中的高度非线性预测。

- scGen的动机:我们可以直接看umap,发现从对照组到刺激组(左),在每个细胞类型(右)中,给人一种非线性的对称感。我们可以用神经网络学习这个关系,从而在新细胞类型上推断扰动后的响应。
scButterfly的扰动和scGen一样,也是在已知细胞类型对照数据和刺激数据上训练,然后预测新细胞类型的扰动响应,并且都是单基因扰动
图增强基因激活与抑制模拟器(GEARS),这是一种将深度学习与基因-基因关系知识图谱相结合的计算方法,用于模拟基因扰动的效应。生物知识的融入使GEARS能够预测单基因或基因组合扰动的结果,即使这些基因或组合此前没有实验扰动数据。在预测来自七个不同数据集的单基因和双基因扰动结果时,GEARS的表现均优于现有方法。此外,GEARS能够检测五种不同的基因相互作用亚型,并通过预测训练中未见过的表型,将预测能力泛化到扰动空间的新区域。因此,GEARS可直接影响未来扰动实验的设计。
GEARS框架
GEARS是一种基于深度学习的模型,可预测组合扰动一个或多个基因(扰动集)后的基因表达结果。给定未受扰动的单细胞基因表达数据以及所应用的扰动集(图1a),其输出为扰动后细胞的转录状态。

- 图1a:给定未受扰动的基因表达(绿色)和施加的扰动(红色),预测基因表达结果(紫色)。每个方框对应一个独立基因,箭头表示表达变化。
GEARS提出了一种新方法,即使用不同的多维嵌入(用于表示有意义概念的任意数字向量;图1b)来表征每个基因及其扰动。在训练过程中,每个基因的嵌入会被调整以表征该基因的关键特征。将表征拆分为两个多维组件,使GEARS能够更充分地捕捉基因特异性的扰动响应异质性。每个基因的嵌入会依次与扰动集中每个基因的扰动嵌入相结合,最终用于预测该基因的扰动后状态。这一预测以一个单一的“跨基因”嵌入向量为条件,该向量捕捉了每个细胞的全转录组信息。
GEARS具有独特的能力,能够预测涉及一个或多个缺乏实验扰动数据基因的扰动集结果。为此,GEARS在学习基因嵌入时采用基因共表达知识图谱整合基因-基因关系先验知识,并在学习基因扰动嵌入时使用基因本体论(GO)衍生的知识图谱。这一方法基于两个生物学直觉:(i)具有相似表达模式的基因可能对外部扰动产生相似响应;(ii)参与相似通路的基因在扰动后可能影响相似基因的表达(图1b)。根据目标基因集的不同,其他知识图谱(如大型上下文特异性网络)可能更适用。GEARS通过图神经网络(GNN)架构将这种基于图的归纳偏置转化为实际功能。

- 图1b:GEARS模型架构(i)对于未扰动状态下的每个基因,GEARS初始化一个基因嵌入向量(绿色)和一个基因扰动嵌入向量(红色)(ii)。这些嵌入向量被指定为基因关系图和扰动关系图中的节点特征(iii)。图神经网络(GNN)用于融合每个图中相邻节点的信息。每个生成的基因嵌入会与扰动集中每个扰动的扰动嵌入相加(iv)。输出通过跨基因层在所有基因间进行组合,并输入特定基因的输出层(v)。最终结果为扰动后的基因表达;MLP为多层感知机。
预测新的生物学表型

- 图a:一组基因的所有成对组合扰动结果的预测流程。
- 图b:用于训练 GEARS 的 102 个单基因扰动和 128 个双基因扰动的扰动后基因表达低维表示。随机选择部分进行了标记。
- 图c:GEARS 对实验中观察到的 102 个单基因的所有 5,151 (一共(102*102-102)/2个) 个成对组合的扰动后基因表达进行预测。预测的扰动后表型(非黑色符号)通常与实验观察到的表型(黑色符号)不同。颜色表示使用标记基因表达标注的 Leiden 簇。
图4b和图4c中的UMAP图基于Norman等人数据集中102个单基因扰动的所有成对组合的GEARS预测扰动后基因表达谱生成。该数据集中共有105个单基因扰动,本图使用了人类物种基因本体数据库中存在的102个基因的扰动数据。图4c展示了包含所有5151种可能的双基因扰动以及102种单基因扰动的完整UMAP扰动后结果。图4b仅使用Norman中用于训练GEARS的扰动(102种单基因扰动和128种双基因扰动)的GEARS预测扰动后基因表达谱绘制。因此,图4b是图4c数据的子集。
聚类使用scanpy中默认参数的Leiden聚类(分辨率=1)进行。图4b和4c中显示的簇使用Norman中的表型标签进行标注。如果任何单个簇或簇组包含Norman等人标注为表现出特定表型的扰动,则整个簇或簇组在图4b或4c中被标注为显示该特定表型。这些图中所有其他簇未赋予表型标签。
每个散点代表一个基因表达谱(比如经过指定扰动的一组细胞的平均表达量)。
scFoundation+GEARS
将基因符号列表统一为19,264个,并在每个数据集上构建了基因共表达网络。参照原始GEARS研究的设计方案:对于单基因扰动实验,随机选取75%的扰动样本作为训练数据;对于双基因扰动实验,训练集仅包含两个基因均属于已知基因集(0/2未见过)的75%组合,其余所有含未知基因的组合(1/2和2/2未见过)均保留作为测试集。随后,通过设置训练周期为15轮、批处理量为30来训练GEARS基线模型。
通过移除scFoundation最后一层MLP,从解码器提取基因上下文嵌入作为共表达网络的节点特征。训练过程中固定scFoundation参数不动,仅对下游GEARS模型进行训练,并采用梯度累积策略以保证与基线模型保持一致的等效批处理规模。

- 在每个数据集中生成共表达网络,用大模型输出的gene token作为网络节点embedding,结合扰动embedding预测转录结果。
与scGPT的不同之处:scGPT没有使用GEARS,是类似scGen的方式。其次,scGPT在每个输入基因的位置附加了一个二元条件标记,用以表明该基因是否受到了扰动。scGPT使用一个对照细胞作为输入,将受扰动的细胞作为目标。这是通过将每个受扰动的细胞与一个未受扰动的对照细胞随机配对来构建输入 - 目标对实现的。因此,该模型学会了基于对照基因的表达情况和扰动标记来预测扰动后的响应。
CellFM的做法与scFoundation一样。
补充内容:化学扰动和基因扰动(参考UniPERT)
下面补充介绍化学扰动和基因扰动的典型架构(参考UniPERT:https://www.biorxiv.org/content/10.1101/2025.02.02.635055v1)
基因扰动:
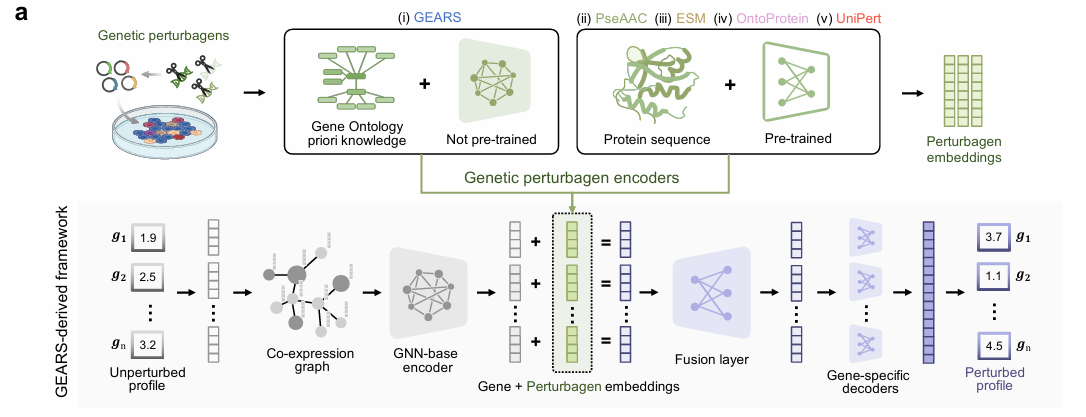
- GEARS-based:GEARS衍生的遗传扰动结果预测框架示意图。给定未扰动的基因表达谱(左下角,即对照组),其中每个基因通过基于基因共表达网络的图神经网络(GNN)进行编码。遗传扰动因子的嵌入表示(左上角)可通过以下方式获得:
1.整合基因本体论(Gene Ontology)先验知识与GNN(如原始GEARS模型)
2.从可扩展的预训练/预定义蛋白质序列表征方法中提取,包括:- ii) PseAAC(伪氨基酸组成)
- iii) ESM(进化-scale模型)
- iv) OntoProtein(基于本体的蛋白质表征)
- v) 本文提出的UniPert模型
- 单一或多个扰动因子的嵌入(绿色)随后被添加到每个基因的嵌入(灰色)中,生成扰动后的基因嵌入(紫色)。这些嵌入向量依次通过融合层和基因特异性解码器,最终转换为预测的扰动后基因表达值。
化学扰动:

- CPA-based:CPA衍生的化学扰动结果预测框架示意图。未扰动谱(左下角)向量被投影到低维潜在空间,而化学扰动因子(左上角),即小分子,使用传统分子指纹特征进行数字化,如i) chemCPA模型,或通过先进表示方法编码,如ii) Uni-Mol、iii) KPGT和iv) UniPert。随后,扰动因子嵌入(黄色)与其他协变量嵌入被添加到潜在对照嵌入(灰色)中,解码后生成预测的扰动后基因谱向量(紫色)。