量子退火实用案例(1):量子退火求解化学中的分子吸附问题,10小时缩短为15分
这篇论文介绍了一种利用量子退火(Quantum Annealing, QA)预测多分子在固体表面吸附的方法,具体研究了CO分子在PdZn (111)表面的吸附行为。论文中使用了密度泛函理论(Density Functional Theory, DFT)的计算结果来估计QUBO(Quadratic Unconstrained Binary Optimization)矩阵,并通过求解QUBO问题来确定最稳定的吸附构型。以下是详细的步骤讲解:
https://pubs.acs.org/doi/10.1021/jacsau.3c00018
研究背景
下图展示了对分子吸附位点优化方法的比较,分为“传统方法”(Conventional methods)和“本文提出的方法”(Proposed method in this work)两大部分。
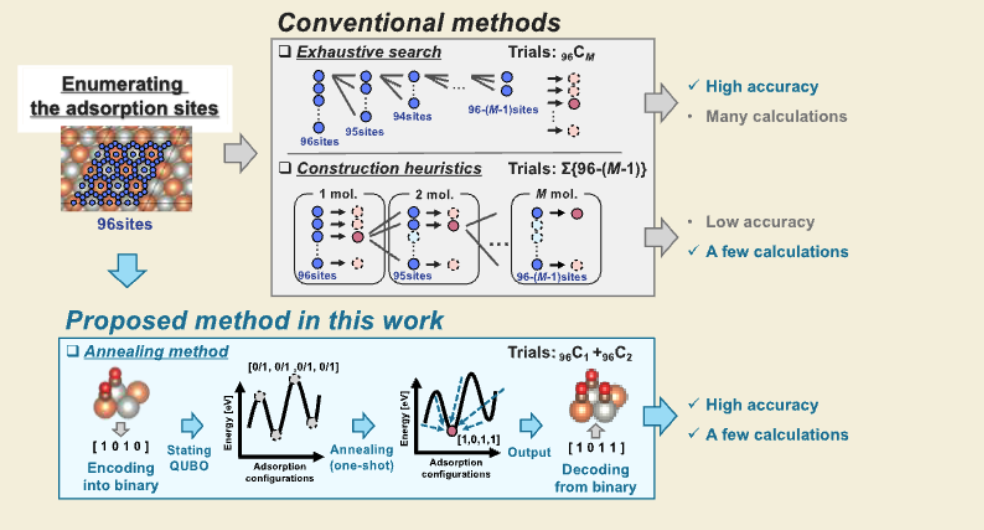
方法对比与意义
- 传统方法:
- 穷举搜索:精度高,但计算量随M呈指数级增长,实际应用中不可行。
- 构造启发式:计算量少,但精度不足,可能无法找到最优吸附构型。
- 退火方法:
- 结合了QUBO模型和退火算法,实现了高精度与低计算量的平衡。
- 计算次数仅为₉₆C₁ + ₉₆C₂(单分子和双分子吸附的组合),效率远高于穷举搜索,同时保持高精度。
- 适用于大规模吸附位点优化,尤其是在高覆盖率或复合材料研究中。
1. QUBO问题的背景与定义
QUBO是一种组合优化问题的数学形式,目标是找到一组二元变量(0或1),使得某个二次函数的值最小化。其目标函数一般表示为:
H = ∑ i , j = 1 N W i j σ i σ j + c \mathcal{H} = \sum_{i,j=1}^N W_{ij} \sigma_i \sigma_j + c H=i,j=1∑NWijσiσj+c
其中:
- σ i \sigma_i σi 是二元变量,取值为0或1;
- W i j W_{ij} Wij 是QUBO矩阵的元素,表示变量之间的相互作用;
- c c c 是一个常数。
在这篇论文中,作者将吸附问题转化为QUBO问题:
- σ i = 1 \sigma_i = 1 σi=1 表示第 i i i 个吸附位点被CO分子占据;
- σ i = 0 \sigma_i = 0 σi=0 表示该位点未被占据;
- H \mathcal{H} H 被定义为体系的总吸附能,目标是找到使 H \mathcal{H} H 最小的 σ i \sigma_i σi 配置,即最稳定的吸附构型。
由于QUBO矩阵是对称的( W i j = W j i W_{ij} = W_{ji} Wij=Wji),且当没有吸附发生时吸附能为0,论文中将常数 c c c 设为0。目标函数可以进一步拆分为对角项和非对角项:
H = ∑ i = 1 N ∑ j > i N 2 W i j σ i σ j + ∑ i = 1 N W i i σ i \mathcal{H} = \sum_{i=1}^N \sum_{j>i}^N 2 W_{ij} \sigma_i \sigma_j + \sum_{i=1}^N W_{ii} \sigma_i H=i=1∑Nj>i∑N2Wijσiσj+i=1∑NWiiσi
这里:
- 对角项 W i i W_{ii} Wii 表示单一分子在第 i i i 个位点吸附时的贡献;
- 非对角项 W i j W_{ij} Wij 表示第 i i i 和第 j j j 个位点同时被占据时的相互作用。
2. 使用DFT计算吸附能
为了构建QUBO矩阵 W i j W_{ij} Wij,需要确定每个元素的具体值。论文中使用了DFT计算来获取这些值,具体分为以下两种情况:
(1) 单分子吸附能(对角元素 W i i W_{ii} Wii)
对于单分子吸附,假设只有一个CO分子吸附在第 k k k 个位点上,则 σ k = 1 \sigma_k = 1 σk=1,其他 σ i = 0 \sigma_i = 0 σi=0。此时,目标函数简化为:
H = W k k \mathcal{H} = W_{kk} H=Wkk
这意味着 W k k W_{kk} Wkk 直接等于单分子在第 k k k 个位点上的吸附能。论文中通过DFT计算每个吸附位点的单分子吸附能 E i E_i Ei,并将其赋值给对角元素:
W i i = E i W_{ii} = E_i Wii=Ei
(2) 双分子吸附能(非对角元素 W i j W_{ij} Wij)
对于双分子吸附,假设CO分子同时吸附在第 k k k 和第 l l l 个位点上,则 σ k = σ l = 1 \sigma_k = \sigma_l = 1 σk=σl=1,其他 σ i = 0 \sigma_i = 0 σi=0。目标函数变为:
H = 2 W k l + W k k + W l l \mathcal{H} = 2 W_{kl} + W_{kk} + W_{ll} H=2Wkl+Wkk+Wll
这里, H \mathcal{H} H 等于双分子吸附能 E k l E_{kl} Ekl,通过DFT计算得到。由于 W k k W_{kk} Wkk 和 W l l W_{ll} Wll 已从单分子吸附能中获得,可以解出非对角元素 W k l W_{kl} Wkl:
W k l = E k l − W k k − W l l 2 W_{kl} = \frac{E_{kl} - W_{kk} - W_{ll}}{2} Wkl=2Ekl−Wkk−Wll
即:
W k l = E k l − E k − E l 2 W_{kl} = \frac{E_{kl} - E_k - E_l}{2} Wkl=2Ekl−Ek−El
这表明,非对角元素 W i j W_{ij} Wij 反映了两个吸附位点之间的相互作用,通过双分子吸附能与单分子吸附能的差值计算得出。
3. 构建QUBO矩阵的步骤
基于上述分析,论文中构建QUBO矩阵的具体步骤如下:
- 确定吸附位点
在PdZn (111)表面上,作者识别出 N = 96 N = 96 N=96 个可能的吸附位点。
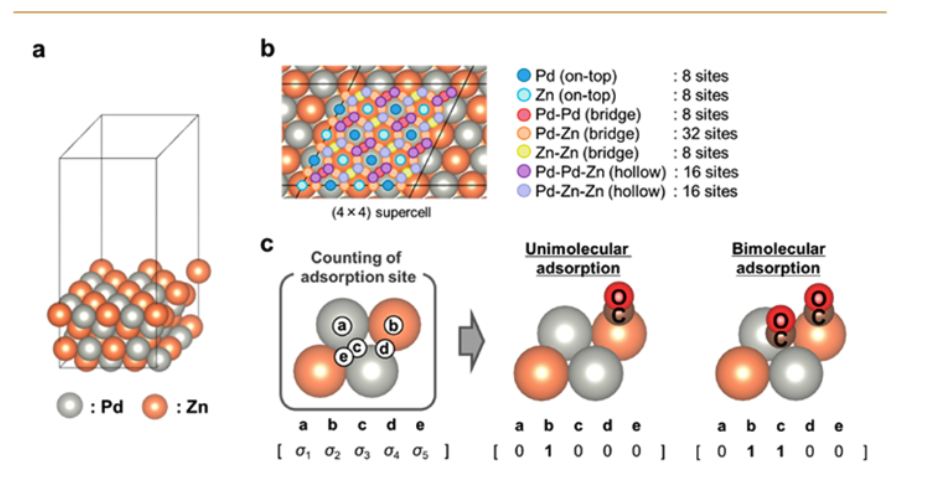
下面详细解释上图。
上图展示了研究中使用的 PdZn 合金表面配置模型,该图由三个部分组成:
部分 (a):PdZn 表面的三维模型
这部分显示了研究中考虑的 PdZn 合金表面的三维结构模型。这是一个(111)晶面的切面,展示了钯(Pd)和锌(Zn)原子在表面上的排列方式。这种表面被选择是因为 PdZn 是一种金属间化合物,与纯金属相比具有较低的对称性,这使得分子吸附行为研究更具挑战性和实际意义。
部分 (b):PdZn 表面上的吸附位点类型和数量
这部分详细标识了 PdZn(111)表面上所有可能的吸附位点类型和数量:
- 图中用不同颜色和标记表示不同类型的吸附位点
- 图例显示包括:
- Pd(on-top):钯原子顶位吸附点(5个)
- Zn(on-top):锌原子顶位吸附点(8个)
- Pd-Pd(bridge):两个钯原子之间的桥位吸附点(4个)
- Pd-Zn(bridge):钯和锌原子之间的桥位吸附点(18个)
- Zn-Zn(bridge):两个锌原子之间的桥位吸附点(15个)
- Pd-Pd-Zn(hollow):由两个钯和一个锌原子形成的三角洞位吸附点(13个)
- Pd-Zn-Zn(hollow):由一个钯和两个锌原子形成的三角洞位吸附点(16个)
- 四叶草型吸附点(17个)
总共确定了96个不同的吸附位点,这些位点构成了QUBO矩阵的变量数量N。
部分 ©:确定二进制变量矩阵的示意图
这部分解释了如何将吸附位点转化为量子退火计算所需的二进制变量:
- 左侧显示吸附位点的编号方案(a、b、c、d、e等)
- 中间展示单分子吸附的情况,其中某个位点(比如位点c)被占据(标记为1),而其他位点未被占据(标记为0)
- 右侧展示双分子吸附的情况,其中两个位点(比如位点a和位点e)同时被占据(都标记为1)
这种二进制表示方法是整个量子退火方法的基础。对于每个吸附位点,变量σᵢ取值为0(表示该位点无分子吸附)或1(表示该位点有分子吸附)。在QUBO问题中,这些二进制变量及其相互作用构成了要优化的能量函数。
通过这种方式,研究人员将化学中的分子吸附问题转化为适合量子退火算法处理的QUBO问题,从而解决了传统方法面临的组合爆炸挑战。
- 计算单分子吸附能
对每个位点 i i i,使用DFT计算单分子吸附能 E i E_i Ei,并将其赋值给QUBO矩阵的对角元素:
W i i = E i W_{ii} = E_i Wii=Ei
#calculating adsorption energy
import glob
import ase
from ase.io import read, write
from pfp_api_client.pfp.calculators.ase_calculator import ASECalculator
from pfp_api_client.pfp.estimator import Estimator
from ase.optimize import BFGSLineSearch
from ase.constraints import FixAtoms, ExpCellFilter, FixedLine
import re
import os
import numpy as np
import pandas as pd
import json
import itertools
import numpy as np
import pandas as pd
from sklearn.linear_model import LinearRegression
fmax = 0.02
mode = 2
mode_index = {2: [True,True], 3: [False, False], 4: [False,True]}
mol_num = [2,1]
adsorption_energy = []
serial = []
serial = 0
model = read(allpath+'/raw/'+'PdZn.cif',format="cif")
estimator = Estimator(calc_mode='CRYSTAL_PLUS_D3')
calculator = ASECalculator(estimator)
model.calc = calculator
PdZn = model.get_potential_energy()
model = read(allpath+'/raw/'+'CO.cif',format="cif")
estimator = Estimator(calc_mode='CRYSTAL_PLUS_D3')
calculator = ASECalculator(estimator)
model.calc = calculator
for num in mol_num:
for serial_num in range(len(glob.glob(allpath+'/Enumerate_{0}mol/*.cif'.format(num)))):
path = allpath+'/Enumerate_{0}mol/{1}.cif'.format(num,serial_num)
model = read(path,format="cif")
estimator = Estimator(calc_mode='CRYSTAL_PLUS_D3',model_version='v2.0.0')
calculator = ASECalculator(estimator)
model.calc = calculator
l = []
for i in model:
l.append(i.position[2])
del i
fix=[]
constraint_list = []
for i in model:
if i.symbol == "Zn" or i.symbol == "Pd":
fix.append(i.index)
else:
constraint_direction = FixedLine(a=i.index, direction=[0, 0, 1])
constraint_list.append(constraint_direction)
constraint_atom = FixAtoms(indices=fix)
constraint_list.append(constraint_atom)
model.set_constraint(constraint_list)
if mode != 2:
constraint = ExpCellFilter(model, hydrostatic_strain=mode_index[mode][0], constant_volume=mode_index[mode][1])
opt = BFGSLineSearch(constraint,logfile="log.txt")
else:
opt = BFGSLineSearch(model,logfile="log.txt")
opt.run(fmax=fmax)
write(path[:-4]+'_opt.cif',model,format="cif")
del l, fix, opt
CO = model.get_potential_energy()
model = read(path[:-4]+'_opt.cif',format="cif")
estimator = Estimator(calc_mode='CRYSTAL_PLUS_D3')
calculator = ASECalculator(estimator)
model.calc = calculator
serial.append(serial_num)
adsorption_energy.append(model.get_potential_energy()-CO*num-PdZn)
df = pd.DataFrame(data={'serial': serial, 'adsorption energy': adsorption_energy})
df.to_csv(allpath+'uni_bi_adsorption_energy.csv')
- 计算双分子吸附能
对每一对位点 ( k , l ) (k, l) (k,l)(由于矩阵对称,只需计算 k < l k < l k<l 的情况),使用DFT计算双分子吸附能 E k l E_{kl} Ekl,然后根据公式计算非对角元素:
W k l = E k l − W k k − W l l 2 W_{kl} = \frac{E_{kl} - W_{kk} - W_{ll}}{2} Wkl=2Ekl−Wkk−Wll
# -*- coding: utf-8 -*-
import numpy as np
from ase.io import read, write
from ase import Atom
from modifiedCatKit import SlabGenerator, get_adsorption_sites, AdsorptionSites
from catkit.gen.symmetry import Symmetry
import pandas as pd
import itertools
import os
import copy
import json
import shutil
class binary_maker():
def __init__(self, example_data, adindex):
self.exdata = example_data
self.adsites = adindex
self.binary_discription = {}
self.adatoms = copy.deepcopy(self.exdata['adatom'])
if len(self.adatoms) == len(set(self.adatoms)):
self.atomdict= {self.adatoms[i]: i for i in range(len(self.adatoms))}
else:
self.double = [atom for atom in set(self.adatoms) if self.adatoms.count(atom) > 1]
self.double_index = [[index for index, _atom in enumerate(self.adatoms) if _atom == atom] for atom in self.double]
self.pseudoadatoms = self.exdata['adatom']
for species in self.double_index:
for order, place in enumerate(species):
self.pseudoadatoms[place] += ('_'*order)
self.atomdict= {self.pseudoadatoms[i]: i for i in range(len(self.adatoms))}
def __call__(self,data):
data = self.adsitecorrespondance(data)
return data
def discription(self):
self.binary_discription = self.adsitecorrespondance_discriptor(self.exdata, self.binary_discription)
return self.binary_discription
def adsitecorrespondance(self, data):
binary_matrix = np.zeros((len(self.adatoms), len(self.adsites)),dtype=int)
try:
for adsite, adatom in zip(data['symmetry_adsorption'].keys().append(data['adindex']), data['symmetry_adsorption'].values().append(data['adatom'])):
for adserial, adsorption in enumerate(adsite):
for adsite in adsorption:
double = [atom for atom in set(adatom) if adatom.count(atom) > 1]
double_index = [[index for index, _atom in enumerate(adatom) if _atom == atom] for atom in double]
pseudoadatoms = copy.deepcopy(adatom)
for species in double_index:
for order, place in enumerate(species):
pseudoadatoms[place] += ('_'*order)
_binary_matrix=copy.deepcopy(binary_matrix)
_binary_matrix[self.atomdict[pseudoadatoms[adserial]]][self.adsites.index(adsite[adserial])] = 1
if len(self.adatoms) == len(set(self.adatoms)):
try:
binary_list.append(_binary_matrix)
except:
binary_list=[_binary_matrix]
else:
permutation = []
for doubles in self.double_index:
permutation.append(list(itertools.permutations(doubles)))
possible_binary_combination = list(itertools.product(permutation))
binary_list = []
for possibility in possible_binary_combination:
indice = np.arange(len(self.adatoms)+1)
for before, after in zip(self.double_index, possibility):
for after_frac in after:
indice[before] = list(after_frac)
binary_list.append(copy.deepcopy(binary_matrix[indice]))
data['binary']['adindex'] = binary_list
except:
for adserial, adsorption in enumerate(data['adindex']):
for adsite in adsorption:
double = [atom for atom in set(data['adatom']) if data['adatom'].count(atom) > 1]
double_index = [[index for index, _atom in enumerate(data['adatom']) if _atom == atom] for atom in double]
pseudoadatoms = data['adatom']
for species in double_index:
for order, place in enumerate(species):
pseudoadatoms[place] += ('_'*order)
binary_matrix[self.atomdict[pseudoadatoms[adserial]]][self.adsites.index(data['adindex'][adserial])] = 1
if len(self.adatoms) == len(set(self.adatoms)):
data['binary']['adsitecorrespondance'] = [binary_matrix]
else:
permutation = []
for doubles in self.double_index:
permutation.append(list(itertools.permutations(doubles)))
possible_binary_combination = list(itertools.product(permutation))
binary_list = []
for possibility in possible_binary_combination:
indice = np.arange(len(self.adatoms))
for before, after in zip(self.double_index, possibility):
for after_frac in after:
indice[before] = after_frac
binary_list.append(copy.deepcopy(binary_matrix[indice]))
data['binary']['adsitecorrespondance'] = np.sum(binary_matrix[indice],axis=0)
return data
def adsitecorrespondance_discriptor(self,data,discription):
caption = 'this method generates a binary which {0} adsorption sites (adsorption site: {1}) repeats {2} times as the number of adsorbent (atom: {3}) plus None'.format((len(self.adatoms)+1)*len(self.adsites), self.adsites, len(self.adatoms)+1, self.adatoms)
discription['adsiteindex'] = caption
return discription
def adsite2POSCAR_symmetry(POSCAR, connectedatoms, out_dir, adoffset=[2.,2.,2.,2.], cutoff=18.0, symmetry_reduced=False, tol=2e-2):
"""
making POSCAR regarding adsorption molecule
input:
POSCAR(str); directory
connectedatoms(np.array); ([atom_index(ex. 'C'),..],...)
out_dir(str); directory
addoffset(list); [2.,2.,2.,2.] as default
cutoff(float); 18.0 as default
symmetry_reduced(bool); False as default
tol(float); 2e-2 as default
dual(bool); False as dafault, wheather adsorption can occur on other adsorbent or not.
output:
nothing but save POSCAR made by this function
"""
pwd = os.getcwd()
if os.path.exists(out_dir):
shutil.rmtree(out_dir)
os.makedirs(out_dir)
tmp = os.chdir(out_dir)
model = read(POSCAR, index=-1, format="cif")
slab_gen = SlabGenerator(model,miller_index=(0,0,1),layers=4,fixed=2,vacuum=None,
standardize_bulk=False,primitive=False,cutoff=cutoff)
slab = slab_gen.get_slab(size=1)
ad_correspondance, adsite_index = adsite_symmetry_set(slab,tol=tol)
coordinates, connectivity, adindices, vector=get_adsorption_sites(slab, symmetry_reduced=False, tol=tol)
addata = []
double_count_adset = []
adsite_index2reduced_adindices, reduced_adindices2adsite_index = adsite_index_correspond_adindices(slab,tol=tol)
for adsorpconfiguration in itertools.permutations(range(len(connectivity.tolist())), len(connectedatoms)):
# Output all combinations of adsorption, and then reduce by considering symmetry
tmp_dict = {}
data_dict = {}
for index in range(len(connectedatoms)):
# tmp_dict{serial of adsorption site: adsorbate molecule}
tmp_dict['{}'.format(adindices[adsorpconfiguration[index]])] = connectedatoms[index]
if not index:
data_dict = {'binary': {}, 'adatom': [connectedatoms[index]], 'coordination': [np.array(coordinates.tolist()[adsorpconfiguration[index]])],
'connectivity': [connectivity.tolist()[adsorpconfiguration[index]]], 'adindex': [adindices[adsorpconfiguration[index]]], 'advector': [np.array(vector.tolist()[adsorpconfiguration[index]])]}
else:
data_dict['adatom'].append(connectedatoms[index])
data_dict['coordination'].append(np.array(coordinates.tolist()[adsorpconfiguration[index]]))
data_dict['connectivity'].append(connectivity.tolist()[adsorpconfiguration[index]])
data_dict['adindex'].append(adindices[adsorpconfiguration[index]])
data_dict['advector'].append(np.array(vector.tolist()[adsorpconfiguration[index]]))
if symmetry_reduced:
data_dict['symmetry_adsorption'] = []
if tmp_dict not in double_count_adset:
double_count_adset.append(tmp_dict)
#Considering symmetry
for symmetry_operation in range(ad_correspondance.shape[0]):
tmp_dict = {}
for index in range(len(connectedatoms)):
operatedsite = ad_correspondance[symmetry_operation,adsite_index.tolist().index(adsite_index2reduced_adindices['{}'.format(adindices[adsorpconfiguration[index]])])]
if operatedsite == -1:
continue
tmp_dict['{}'.format(reduced_adindices2adsite_index['{}'.format(adsite_index[operatedsite])])] = connectedatoms[index]
if tmp_dict not in double_count_adset:
double_count_adset.append(tmp_dict)
data_dict['symmetry_adsorption'].append(tmp_dict)
addata.append(data_dict)
else:
addata.append(data_dict)
binary_creater = binary_maker(copy.deepcopy(addata[0]), get_adsorption_sites(slab, symmetry_reduced=False, tol=tol)[2])
binary_discription = binary_creater.discription()
for serial, addict in enumerate(addata):
surface = copy.deepcopy(slab)
for adsorbent_num in range(len(addict['adatom'])):
C_position=addict['coordination'][adsorbent_num]+addict['advector'][adsorbent_num]*adoffset[addict['connectivity'][adsorbent_num]]
surface += Atom(addict['adatom'][adsorbent_num],C_position)
surface += Atom('O',C_position+(0,0,1.18))
write('./{}.cif'.format(serial),surface,format="cif")
addict['serial'] = serial
addata[serial] = binary_creater(copy.deepcopy(addict))
adlist = []
for data in addata:
tmp_dict = {}
bin_dict = {}
for key in data['binary'].keys():
bin_dict[key] = [copy.deepcopy(data['binary'][key][0]).tolist()]
tmp_dict['binary'] = bin_dict
tmp_dict['adatom'] = copy.deepcopy(data['adatom'])
tmp_dict['coordination'] = [copy.deepcopy(data['coordination'][0]).tolist()]
tmp_dict['connectivity'] = copy.deepcopy(data['connectivity'])
tmp_dict['adindex'] = copy.deepcopy(data['adindex'])
tmp_dict['advector'] = [copy.deepcopy(data['advector'][0]).tolist()]
tmp_dict['serial'] = copy.deepcopy(data['serial'])
adlist.append(tmp_dict)
with open('adsorption_data.json', 'ab+') as f:
f.write(json.dumps([{'addata': adlist}]).encode())
tmp = os.chdir(pwd)
return addata, binary_discription, adindices
def adsite_symmetry_set(slab,tol=2e-2):
sym = Symmetry(slab,tol=tol)
match=AdsorptionSites(slab,tol=tol)
rotations, translations = sym.get_symmetry_operations(affine=False)
rotations = np.swapaxes(rotations, 1, 2)
affine = np.append(rotations, translations[:, None], axis=1) #symmetric operation(rotations and translations,xyz)
points = match.frac_coords #Creating relative coordinates of adsorption sites after expanding the supercell(adsorption sites position,xyz)
true_index = match.get_periodic_sites(False)
affine_points = np.insert(points, 3, 1, axis=1)
operations = np.dot(affine_points, affine) #relationship between symmetric operation and adsorption sites position,xyz)
symmetry_match = np.arange(points.shape[0])
adsite_index = match.get_periodic_sites()
original_adsite = match.get_periodic_sites()
ad_correspondance = np.zeros([operations.shape[1],original_adsite.shape[0]],dtype="int8")-1
for i, j in enumerate(symmetry_match):
if i != j:
continue
d = operations[i, :, None] - points #Comparing the coordinates after each symmetry operation with all adsorption points
d -= np.round(d) #Extraction of fractional part
dind = list(zip(*np.where((np.abs(d) < match.tol).all(axis=2))))
for sym_operation, adsite in dind:
if adsite not in original_adsite:
continue
else:
ad_correspondance[sym_operation,np.where(adsite_index==true_index[i])] = np.where(adsite_index==true_index[adsite])
return ad_correspondance, adsite_index
def adsite_index_correspond_adindices(slab,tol=2e-2):
sites = AdsorptionSites(slab, surface_atoms=None, tol=tol)
s = sites.get_periodic_sites()
adsite_index2reduced_adindices={}
reduced_adindices2adsite_index={}
for frac in s:
reduced_indices = []
for reduced_frac in sites.r1_topology[frac]:
reduced_indices.append(sites.index[reduced_frac])
adsite_index2reduced_adindices["{}".format(reduced_indices)]=frac
reduced_adindices2adsite_index["{}".format(frac)]=reduced_indices
return adsite_index2reduced_adindices, reduced_adindices2adsite_index
- 构造完整QUBO矩阵
通过上述计算,填充 N × N N \times N N×N 的QUBO矩阵。总共需要 N N N 次单分子吸附计算和 ( N 2 ) = N ( N − 1 ) 2 \binom{N}{2} = \frac{N(N-1)}{2} (2N)=2N(N−1) 次双分子吸附计算。对于 N = 96 N = 96 N=96,这是 96 + 4560 = 4656 96 + 4560 = 4656 96+4560=4656 次DFT计算。虽然数量较多,但远小于直接计算多分子吸附配置的组合数(如 ( 96 3 ) = 142880 \binom{96}{3} = 142880 (396)=142880)。
def create_QUBO(energy_csv, data, adindices, barrier=500):
df = pd.read_csv(energy_csv)
serials = []
QUBO = np.full((len(np.array(data[0]['binary']['adsitecorrespondance']).flatten()),len(np.array(data[0]['binary']['adsitecorrespondance']).flatten())),barrier,dtype= float)
for row in range(len(df)):
serials.append(df.iat[row,1])
#print(serials)
for addata in data:
#print(addata['serial'])
if addata['serial'] in serials:
adpointlist=np.where((np.array(addata['binary']['adsitecorrespondance']).flatten())==1)[0]
#print(adpointlist)
if len(adpointlist) == 2: #bimolecular adsorption
QUBO[adpointlist[1],adpointlist[0]]=df.iat[serials.index(addata['serial']),2]
QUBO[adpointlist[0],adpointlist[1]]=df.iat[serials.index(addata['serial']),2]
else: #unimolecular adsorption
QUBO[adpointlist[0],adpointlist[0]]=df.iat[serials.index(addata['serial']),2]
for symmetry in addata['symmetry_adsorption']:
if len(symmetry.keys()) == 2:
adpoint_sym_0=list(map(int,list(symmetry.keys())[0][1:-1].split(',')))
adpoint_sym_1=list(map(int,list(symmetry.keys())[1][1:-1].split(',')))
QUBO[adindices.index(adpoint_sym_0),adindices.index(adpoint_sym_1)]=df.iat[serials.index(addata['serial']),2]
QUBO[adindices.index(adpoint_sym_1),adindices.index(adpoint_sym_0)]=df.iat[serials.index(addata['serial']),2]
else:
adpoint_sym_0=list(map(int,list(symmetry.keys())[0][1:-1].split(',')))
QUBO[adindices.index(adpoint_sym_0),adindices.index(adpoint_sym_0)]=df.iat[serials.index(addata['serial']),2]
delindex = np.where(np.sum(QUBO,axis=0)==barrier*len(np.array(data[0]['binary']['adsitecorrespondance']).flatten()))
if len(delindex)>0:
_QUBO = copy.deepcopy(QUBO)
del QUBO
QUBO = np.delete(np.delete(_QUBO,list(delindex),0),list(delindex),1)
del _QUBO
for raw in range(QUBO.shape[0]):
for column in range(raw+1,QUBO.shape[0]):
QUBO[raw][column] = (QUBO[raw][column]-(QUBO[raw][raw]+QUBO[column][column]))/2
QUBO[column][raw] = (QUBO[column][raw]-(QUBO[raw][raw]+QUBO[column][column]))/2
return QUBO, delindex
4. 求解QUBO问题
构建好QUBO矩阵后,论文使用量子退火方法求解优化问题,具体步骤如下:
(1) 设置约束条件
目标是预测 M M M 个CO分子吸附时的最优配置。论文中考虑了两种约束:
-
初始尝试:等式约束
要求恰好 M M M 个位点被占据,即 ∑ i = 1 N σ i = M \sum_{i=1}^N \sigma_i = M ∑i=1Nσi=M。但在高覆盖率时,这无法反映分子解吸现象。 -
改进:不等式约束
考虑到高覆盖率下可能发生解吸,改为 ∑ i = 1 N σ i ≤ M \sum_{i=1}^N \sigma_i \leq M ∑i=1Nσi≤M。这允许吸附分子数小于 M M M,更符合实际情况。约束通过退火求解器直接处理,或通过在目标函数中添加罚项实现。
#Making QUBO
energy_csv = allpath+'uni_bi_adsorption_energy.csv'
original_QUBO, delindex= Toolkit.create_QUBO(energy_csv=energy_csv, data=addata_sum, adindices=adindice_2mol, barrier=500)
import json
with open(allpath+'QUBOmartrix.json', 'w') as f:
json.dump({'QUBO': original_QUBO.tolist()},f,indent=4)
(2) 使用退火求解器
论文中使用了Fujitsu提供的DA3(Digital Annealer Ver. 3),一种受量子退火启发的经典计算求解器。它能够高效求解QUBO问题,找到使 H \mathcal{H} H 最小的 σ \sigma σ 配置。求解过程包括:
- 输入QUBO矩阵 W i j W_{ij} Wij 和约束条件;
- 运行退火算法,输出最优的 σ = [ σ 1 , σ 2 , … , σ N ] \sigma = [\sigma_1, \sigma_2, \ldots, \sigma_N] σ=[σ1,σ2,…,σN]。
#DA
from python_fjda import fujitsu_json, fjda_scheme, fjda_client
import time
import pandas as pd
#parameter
DAresultpath=allpath+'DAresult.json'
max_adnum=16
trials=3
import json
DAresult = {"mol_serial":["cost", ["state"],["index"],["adsite"],"api_time"]}
with open(DAresultpath, 'w') as f:
json.dump(DAresult, f, indent=4)
for adnum in range(1,max_adnum+1):
trial_dict = {}
for trial in range(trials):
#DA annealing
fj_data=Toolkit.Make_DAinput(adnum=adnum, QUBO=original_QUBO*10000, const=0, desorption=True)
fj_qubo_request = fujitsu_json.QuboRequest_fromdict(fj_data)
dac = fjda_client.fjda(server="dau2-05-1.da.labs.fujitsu.com")
if 1:
qs = dac.qstat()
qa = dac.qstatAsync()
#print('Use fjda-server', dac.fjda_server)
fj_res = dac.v3_qubo_solve(basic_request=fj_qubo_request)
while True:
time.sleep(10)
fj_res2 = dac.get_results(fj_res.id)
if fj_res2.status in ('completed', 'deleted', 'error'):
break
#Output
r = fj_res2.results
r.cost_min[np.where(r.penalty_min!=0)] = 10e10
arg = np.argmin(np.array(r.cost_min))
df = pd.DataFrame({'state': np.where(np.array(r.state_min[arg])==1),
'cost': r.cost_min[arg],
'penalty': r.penalty_min[arg]})
trial_dict[r.cost_min[arg]]=[df,trial,r.state_min[arg],r._api_time]
min_arg = sorted(trial_dict.items())[0][0]
print('number of molecule:',adnum,'trial:',trial_dict[min_arg][1])
print(trial_dict[min_arg][0])
#Data
position_list=[]
element_list=[]
for element in range(len(adindice_1mol)):
if list(map(int,trial_dict[min_arg][2]))[element] == 1:
position_list.append(adindice_2mol[element])
element_list.append(element)
with open(DAresultpath) as f:
DAresult=json.load(f)
#list(trial_dict[min_arg][0]['state'])),
DAresult['{0}_{1}'.format(adnum,trial)] = [int(trial_dict[min_arg][0]['cost']),element_list,position_list,trial_dict[min_arg][3]]
with open(DAresultpath, 'w') as f:
json.dump(DAresult, f, indent=4)
(3) 计算吸附能并验证
对于找到的最优配置 σ \sigma σ,通过 H \mathcal{H} H 计算预测的吸附能,并与DFT计算的实际吸附能进行比较。论文中的图4显示,二者吻合良好,验证了方法的准确性。
#Output of DA result as cif file
connectedatoms_1mol_nosym=['C']
outpath_1mol_nosym=allpath+'Enumerate_1mol_nosym/'
addata_1mol_nosym, binary_discription_1mol_nosym, adindice_1mol_nosym= Toolkit.adsite2POSCAR_symmetry(slab_path, connectedatoms=connectedatoms_1mol_nosym, out_dir=outpath_1mol_nosym, adoffset=[2,2,2,2], cutoff=38.0, symmetry_reduced=False, tol=2e-2)
DAresultpath=allpath+'DAresult.json'
cifsavepath=allpath+'annealing/'
output_cif=Toolkit.Make_COadmodel(max_adnum=16,trials=3,unimolpath=outpath_1mol_nosym, DAresultpath=DAresultpath, cifsavepath=cifsavepath)
5. 方法的优势
-
避免组合爆炸
传统方法直接计算多分子吸附的能量需要 ( N M ) \binom{N}{M} (MN) 次DFT计算,计算量随 M M M 指数增长。而该方法仅需 N + ( N 2 ) N + \binom{N}{2} N+(2N) 次DFT计算(对于 N = 96 N=96 N=96 是4656次),大大减少了计算成本。 -
高效预测
QUBO矩阵只需构建一次,随后可用于不同 M M M 的预测。退火求解器能在短时间内(如16分子时132秒)找到最优配置,相比传统方法(38601秒)效率显著提高。 -
准确性
通过仅使用单分子和双分子吸附能,方法能够准确预测多分子吸附的能量和构型,适用于高覆盖率场景。
总结
该论文通过以下步骤使用DFT计算结果估计QUBO矩阵并求解:
- 用DFT计算每个位点的单分子吸附能 E i E_i Ei,设 W i i = E i W_{ii} = E_i Wii=Ei;
- 用DFT计算每对位点的双分子吸附能 E i j E_{ij} Eij,计算 W i j = E i j − E i − E j 2 W_{ij} = \frac{E_{ij} - E_i - E_j}{2} Wij=2Eij−Ei−Ej;
- 构造QUBO矩阵 W i j W_{ij} Wij;
- 在约束 ∑ i = 1 N σ i ≤ M \sum_{i=1}^N \sigma_i \leq M ∑i=1Nσi≤M 下,使用退火求解器找到使 H \mathcal{H} H 最小的 σ \sigma σ 配置。
这种方法将DFT的精确性与量子退火的优化能力结合,为多分子吸附研究提供了一种高效、准确的工具。
代码都是作者提供的整体代码中摘出来的,主要用于逻辑整理。