Cell子刊 IF48.8 | 绘制 949 种癌细胞蛋白质组全景图,AI 算法锁定千种潜在药物靶点
期刊名称:Cancer Cell
影响因子:48.8
期刊分区:JCR 1区/中科院1区
发表时间:2022年8月
作者单位:悉尼大学、威康桑格研究所等
相关疾病:多种癌症
样本数量:949种细胞系、6次重复
样本类型:癌细胞系
01前言

今天小谱给大家带来一篇于2022年7月发表在杂志Cancer Cell(IF=48.8)上的文章,标题为“Pan-cancer proteomic map of 949 human cell lines”。
该文亮点如下:
1、研究团队通过对 949 种人类癌细胞系的蛋白质组进行分析,构建了目前最大的癌细胞系蛋白质组数据集。
2、通过深度学习整合蛋白质组、药物反应和基因编辑数据,发现 70% 癌症易感性标志物仅在蛋白质层面显著。
3. 证实蛋白质网络共调控程度远超基因表达网络,随机选取 1,500 个蛋白仍保留 88% 药物反应预测能力,为临床小样本检测提供理论支撑。
02研究背景
精准医学的核心在于识别特定的生物分子,以此为基础指导治疗方案的选择,从而实现对疾病的精准打击。当前,癌症生物标志物的发现主要依赖功能遗传学与药理学筛选,但由于癌症的复杂性,基因组学往往无法准确预测治疗响应。
随着质谱技术的飞速发展,大规模、可重复的蛋白质组学分析技术已经成熟。本文定量了949个细胞系中的8,498种蛋白质,构建了ProCan-DepMapSanger数据集。该数据集与癌症依赖性图谱进行了整合,还通过深度学习算法,成功识别了一系列与癌症敏感性相关的生物标志物。其中有些是无法通过基因组学或转录组学单独识别的,凸显了蛋白质组学在癌症研究中的独特价值。同时发现蛋白质组在预测癌症表型方面的能力与转录组相当,甚至在某些方面更具优势。
03研究样本
949种来自不同组织的人类细胞系,细胞来源于公共数据库以及私人收藏。
04实验设计
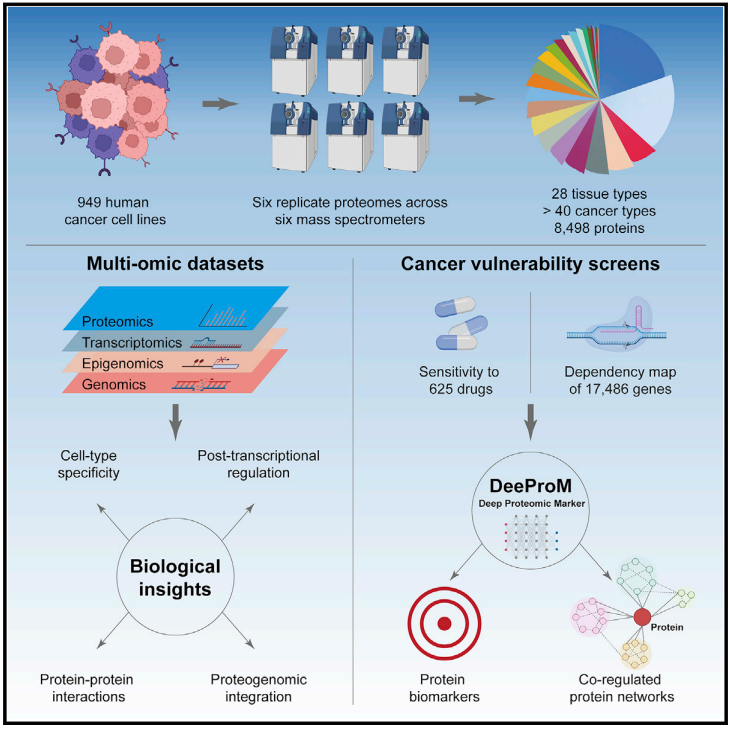
图1 研究流程
对来自超过40种类型的949种人类癌症细胞进行DIA定量,涵盖28种组织和40多种组织学不同的癌症类型,以及广泛的基因型。对每个细胞系的检测进行 6 次技术重复,共定量了8,498 种蛋白质。构建了一个全面的蛋白质组图谱(ProCan-DepMapSanger数据集)。深入探讨了不同类型癌细胞中的转录后调控因素和癌细胞共调节网络。并通过多组学分析,揭示了转录后调控和蛋白质共调节网络的复杂性。
05实验结果
(1)949个癌细胞系蛋白质组的资源
研究结果显示,对来自28种组织和超过40种遗传和组织学上不同的癌症类型的949种人类癌细胞系的蛋白质组,进行了定量并构建了泛癌症蛋白质图谱。该图谱显著拓展了癌症的分子表征与抗癌药物的筛选范围。同时对每组重复、癌症类型、组织类型、批次和仪器等进行皮尔森相关性分析。这些结果表明构建泛癌症蛋白质组学的实验过程是可靠的。
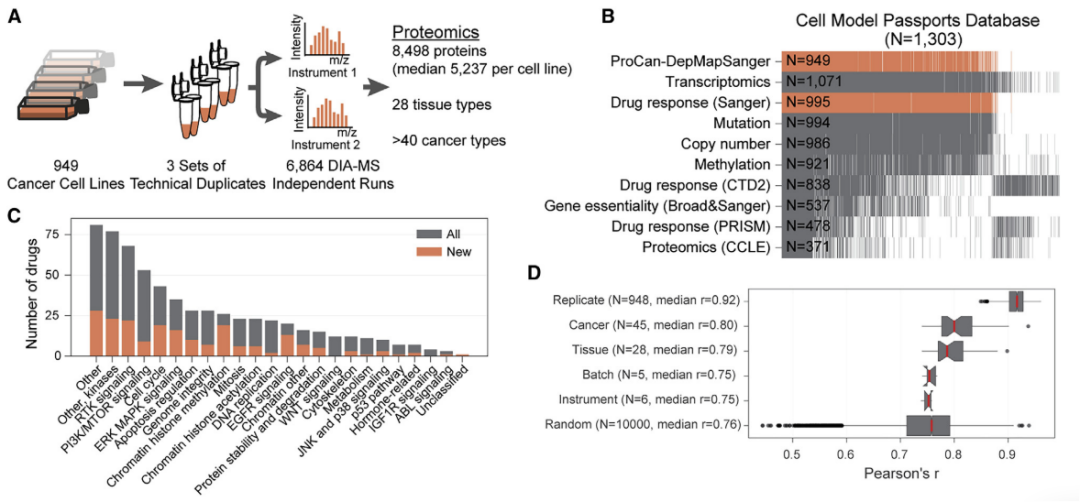
图2 949个人类癌细胞系的泛癌蛋白质组图谱
(2)蛋白质组学图谱揭示细胞起源类型
通过均匀流形近似与投影(UMAP)技术对蛋白质组数据进行降维分析,结果显示,不同组织来源的细胞系(如造血和淋巴细胞、皮肤细胞等)形成了明显的聚类。研究定义了 279 种细胞类型富集蛋白(cell type-enriched proteins),这些蛋白在特定组织中高表达(如淋巴细胞活化相关蛋白、神经元投射蛋白、色素沉着相关蛋白),且比其他蛋白在转录组与蛋白质组间的相关性更高(中位数 r=0.7-0.8)。例如,造血 / 淋巴样细胞系进一步按谱系(如 T 淋巴细胞、B 淋巴细胞、髓系细胞)聚类,显示蛋白质组能有效反映细胞分化状态。
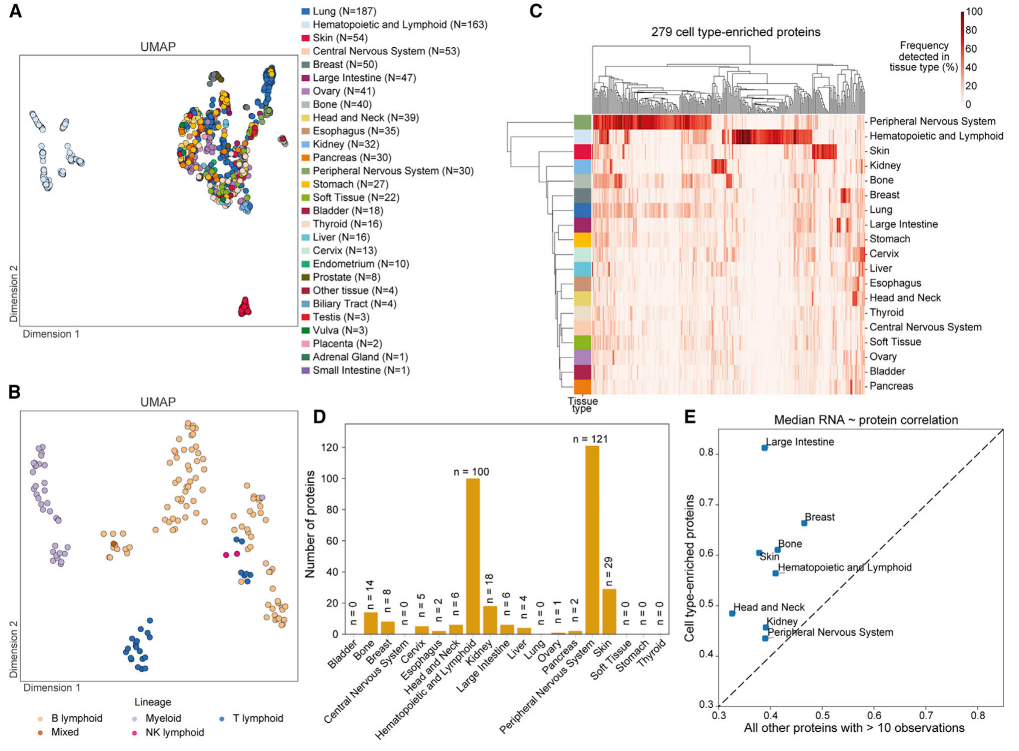
图3 根据细胞类型的不同蛋白质组图谱
(3)不同类型癌细胞中的转录后调控
研究者们使用多组学因子分析(MOFA)整合蛋白质组数据与其他分子(启动子甲基化、基因表达和蛋白质丰度)及表型(药物反应)数据集,以探究癌症细胞系中蛋白质表达模式的驱动因素。MOFA基于贝叶斯群体因子分析框架,能够无监督地整合数据集,推断出一组因子(潜在变量),这些因子可以解释数据中的生物学和实验技术变异性。
分析发现,上皮-间质转化(EMT)标志物vim和E-cadherin与前两个因子相关,解释了数据中大部分的变异性。且EMT标志物在转录组学和蛋白质组学之间表现出高度一致性,对于研究EMT相关的生物学过程和疾病机制具有重要意义。然而,从更广泛的层面来看,蛋白质和转录测量之间的相关性在不同基因之间存在差异,整体相关性并不强,中位数蛋白质-转录组皮尔逊相关系数为0.42。这表明蛋白质组数据能够捕获转录后调控和蛋白质稳态网络的影响。而基因拷贝数与蛋白质水平之间的相关性比与基因表达的相关性更弱。研究还发现,某些蛋白质的表达水平与基因突变状态密切相关,这些蛋白质在癌症细胞的生长和存活中起着关键作用。这些结果表明,转录后调控在不同癌症细胞类型中具有广泛的调控作用,对理解癌症的分子机制具有重要意义。
(4)癌细胞的共调节蛋白质网络
研究还发现蛋白质组测量比转录组和CRISPR-Cas9基因必需性更能揭示已知的蛋白质-蛋白质相互作用(PPIs)。在所有资源中,蛋白质组测量检测已知PPIs的能力更强(召回率曲线下面积[AUC]为0.55-0.80),而转录组(AUC为0.53-0.72)和CRISPR-Cas9基因必需性(AUC为0.51-0.63)的检测能力较弱。例如,EEF2-EIF3I、RPSA-SERBP1和CCT6A-EEF2等蛋白质对之间存在强相关性。这些蛋白质虽然没有被报告直接相互作用,但在高可信度的STRING蛋白质相互作用网络中密切相关。在CRISPR-Cas9基因必需性数据集中,具有较高正蛋白质-蛋白质相关性的蛋白质对癌细胞的存活更为重要。这与它们的转录和蛋白质表达水平增加以及它们参与的途径数量有关。
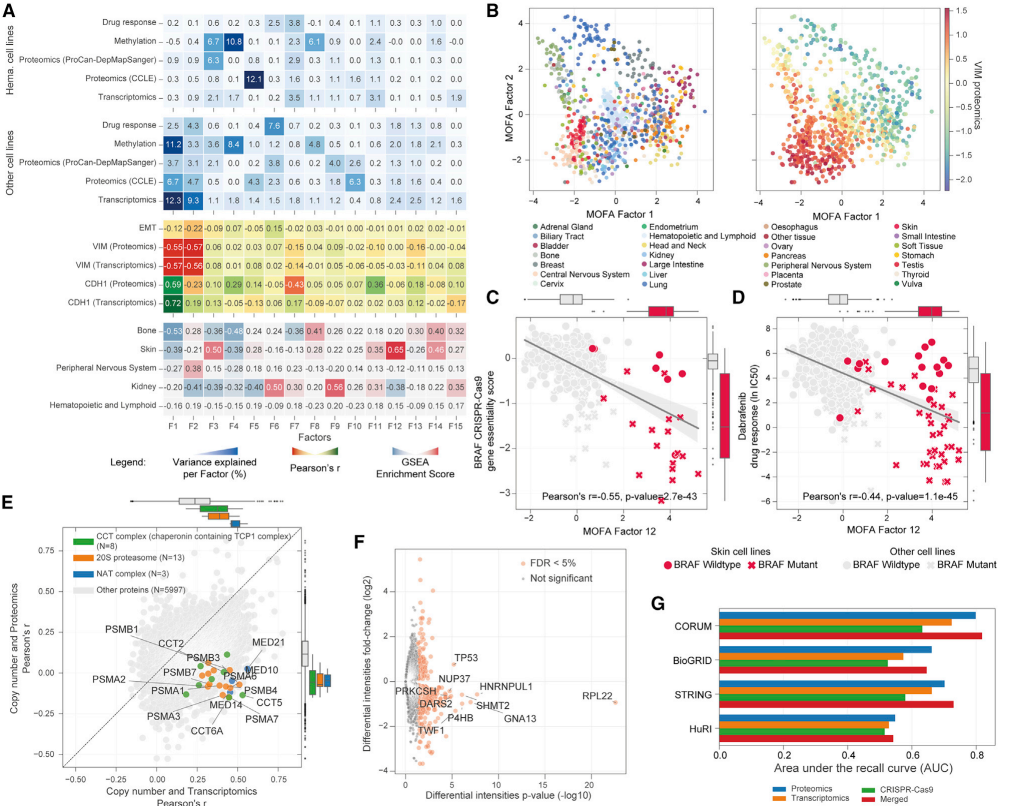
图4 癌细胞系的转录后调控机制
(5)识别癌症易感性的生物标志物
研究团队为预测癌细胞系对药物反应以及CRISPR-Cas9基因必要性的蛋白质生物标志物,使用线性回归来测试蛋白质,药物敏感性和CRISPR-Cas9基因依赖性之间的关联。在最强的显著相关性(FDR < 5%)中,观察到57种药物与其典型靶点的蛋白丰度相关,包括EGFR蛋白丰度与其抑制剂吉非替尼之间的负相关性,以及MET蛋白丰度与其抑制剂药物反应之间的负相关性。同时观察到ERBB2和拉帕替尼之间存在显著的负相关性。大多数显著的药物-蛋白质靶点关联显示负效应,表明当药物靶点更丰富时,细胞系对药物更敏感。之后在蛋白质水平上观察到了与报告的基因拷贝数改变的相关性。
为了识别蛋白质组所特有的生物标志物关联,并且不能单独通过基因表达测量来预测,研究团队开发了一种基于深度学习的计算流程,称为深度蛋白质组学标记(DeeProM)。
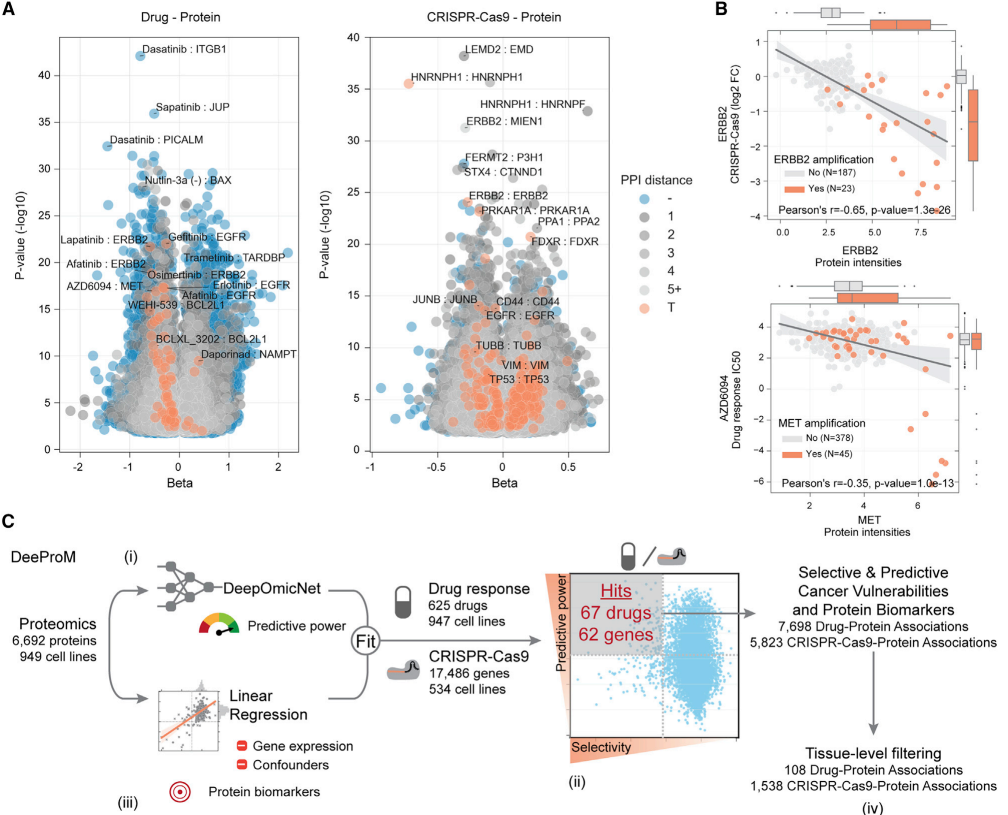
图5 癌症脆弱性的生物标志物
DeeProM评估了所有的药物-蛋白质(n = 4,218,788)和CRISPR-蛋白质(n = 86,584,537)关联,以筛选在细胞系亚群中同时具有良好预测和选择性的癌症敏感性标志物。通过 DeeProM 筛选出 67 种药物和 62 个基因必需性的生物标志物,其中 7,698 个药物-蛋白关联和 5,823 个 CRISPR-蛋白关联在蛋白质水平显著优于转录组模型。例如,ERBB2蛋白丰度与拉帕替尼的反应显著负相关,这与之前在乳腺癌患者来源异种移植模型中的研究一致。此外,DeeProM还鉴定出108个组织特异性的药物-蛋白质关联,这些关联在转录组水平未被发现。这些结果表明,蛋白质组数据能够揭示新的癌症生物标志物,这些生物标志物在转录组水平无法检测到。
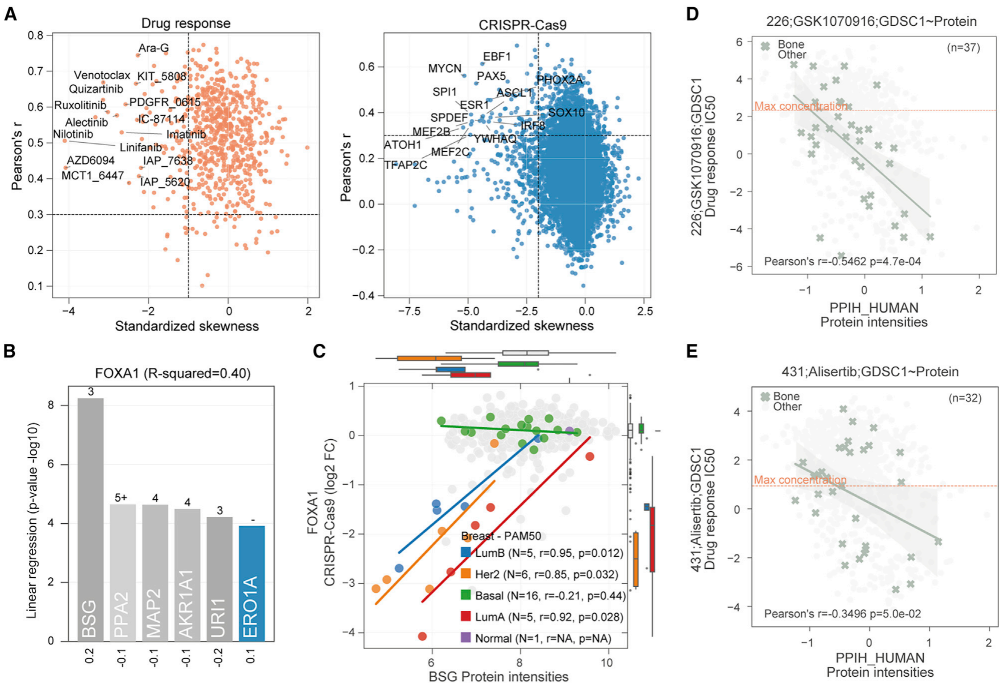
图6 DeeProM鉴定的蛋白质生物标志物
(6)蛋白质子网络对癌细胞表型的预测能力
研究团队评估了蛋白质组和转录组数据在预测药物反应和CRISPR-Cas9基因必需性方面的预测能力。结果表明,蛋白质组和转录组数据的预测能力在训练模型时表现出相似的准确性。随机下采样分析显示,仅 1,500 个随机选择的蛋白质即可保留 88% 的预测能力(r=0.43 vs. 完整数据的 r=0.49),表明蛋白质网络高度互联且冗余。进一步分析蛋白质丰度频率发现,高频蛋白(存在于≥90% 细胞系)在预测中表现最优,在STRING蛋白质相互作用网络中的连接度显著更高。这些结果表明,即使测量蛋白质组的一小部分,也足以表征参与关键细胞表型的基本元素。这是因为蛋白质组中的蛋白质被组织成具有互相关联亚基的复合体和途径,这些复合体和途径在介导关键细胞表型中起着重要作用。
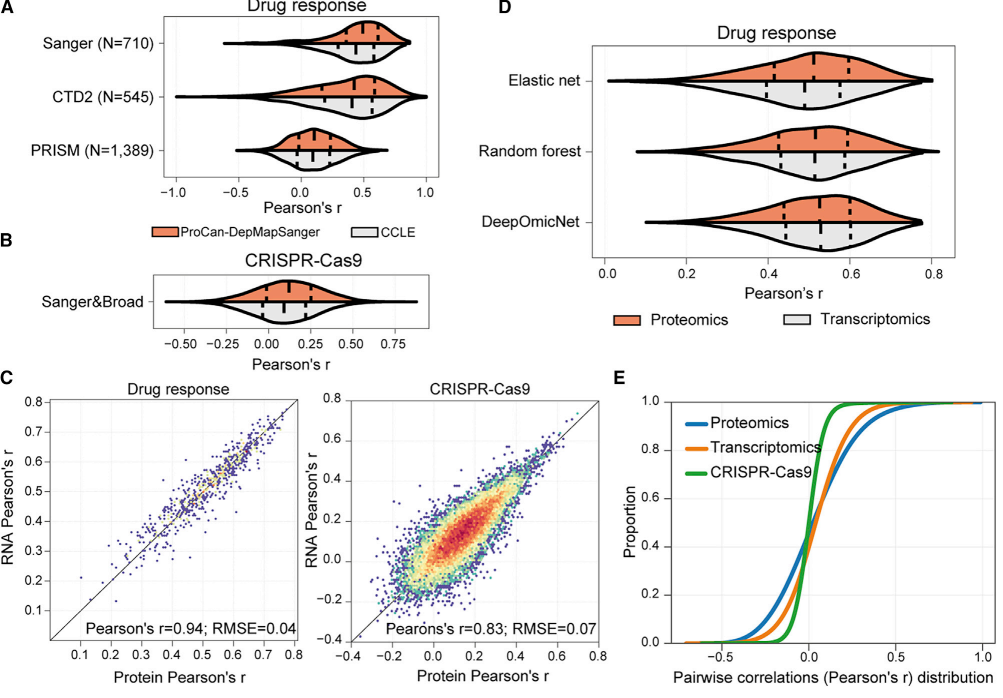
图7 评估DeepOmicNet对多组学数据集的预测能力
小谱评价
本研究通过对949种人类癌细胞系的定量蛋白质组学分析,构建了目前最大的癌细胞系蛋白质组数据集ProCan-DepMapSanger,为癌症研究提供了丰富的资源。并开发了基于深度学习的DeeProM流程,实现了蛋白质组数据与药物反应和CRISPR-Cas9基因必需性筛选的完整整合。
研究结果证实了蛋白质组学在疾病研究中的独特价值。蛋白质水平和转录水平之间的整体相关性并不强,而蛋白质组数据更能够捕获转录后调控和蛋白质稳态网络的影响,且某些蛋白质的表达水平与基因突变状态密切相关,表明了蛋白质水平的研究对于理解癌症的分子机制具有重要意义。
此外,研究还凸显了蛋白质组学技术在生物标志物发现中的重要性。一方面,DeeProM流程能够挖掘出仅在蛋白质水平显著的癌症生物标志物,这些标志物在转录组水平无法被关注到。另一方面,由于蛋白质网络高度互联且冗余,即使是相对较少的蛋白质组成的随机子集就足以反映许多基本的细胞过程,具有良好的预测能力。