【AI4S】基于分子图像与分子描述符的药物筛选预测模型
基于分子图像与分子描述符的药物筛选预测模型
- 引言
- 方法
- 数据集的筛选和处理
- 计算分子描述符(MDs)
- 数据集划分为训练集与测试集,并通过化学空间分析进行验证
- DeepSnap
- DeepLearning
- 基于MD构建预测模型
- 模型综合与评估
- 结果
- 数据分离和化学空间分析
- DeepSnap-DL 模型构建
- MD-based 方法和模型
- 集成和共识模型的优势
- 图表解读
- 创新点
- 结论
- 预测模型的优化
- 动物实验的替代方案
- 中枢神经系统药物分布
- 数据集与性能
- 实用性与局限性
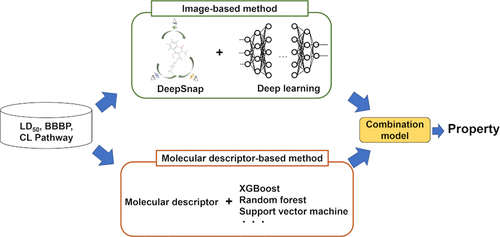
在药物发现阶段,需要开展多种毒性和药代动力学评估作为筛选实验。目前,为减少动物实验的使用及降低研发成本,人们迫切希望开发基于定量构效关系分析的高性能预测模型。为此,我们选取了半数致死剂量(LD50)、血脑屏障穿透性(BBBP)以及清除率(CL)这三项评价指标,并利用636至11,886个化合物构建了针对每项指标的预测模型。首先,我们采用DeepSnap深度学习(DL)方法,以化合物的图像特征构建预测模型。结果显示,该方法对LD50、BBBP和CL途径的预测曲线下面积(AUC)和平衡准确率(BAC)分别达到0.887和0.818、0.893和0.824,以及0.883和0.763。随后,我们利用Molecular Operating Environment、alvaDesc和ADMET Predictor计算了化合物的分子描述符(MDs),并基于这些MDs,通过DataRobot平台构建了相应的预测模型。结果表明,该方法对LD50、BBBP和CL途径的AUC和BAC分别达到0.931和0.805、0.919和0.849,以及0.900和0.807。

论文地址:https://pubmed.ncbi.nlm.nih.gov/37841172/
在本研究中,我们进一步整合了DeepSnap-DL方法与基于MD的方法,构建了集成模型。通过计算两种方法预测概率的平均值,我们得到了一组性能更优的集成模型,其对LD50、BBBP和CL途径的AUC和BAC分别达到0.942和0.842、0.936和0.853,以及0.908和0.832,显示出优于单一方法的综合预测能力。此外,我们还构建了共识模型,仅纳入两种方法结果一致的化合物进行建模。结果显示,这些共识模型对LD50、BBBP和CL途径的BAC分别高达0.916、0.918和0.847,明显高于对应的集成模型,表明其预测性能全面超越了集成模型。综上所述,结合DeepSnap-DL方法与基于MD的方法所构建的预测模型,在LD50、BBBP和CL途径等药物筛选目标上均表现出优异的预测性能。因此,这一方法有望加速药物发现进程,广泛应用于各类药物研发的筛选环节。
引言
为了减少动物实验的使用及研发成本,近年来,基于定量构效关系(QSAR)分析的预测模型已在药物发现阶段得到积极应用。QSAR分析是一种以化合物的分子描述符(MDs)和指纹等特征为基础的方法,目前已成功应用于支持向量机、随机森林、人工神经网络、k近邻法、XGBoost以及深度学习(DL)等多种算法中。在药物发现阶段,QSAR分析主要用于预测药物的药理活性、药代动力学参数及毒性参数。此前,已有多个基于QSAR分析的预测模型被报道,分别针对与毒性相关的参数,如人Ether-a-go-go相关基因的抑制作用和半数致死剂量(LD50),以及与药代动力学相关的参数,包括代谢稳定性、蛋白结合率、血细胞分布、膜通透性、清除率(CL)及其具体清除途径等。因此,这些预测模型已被多家制药公司广泛用于虚拟筛选,以缩小候选化合物范围,并为后续实验提供优先级排序。
目前,针对多种毒性参数和药代动力学参数,已有多项预测模型问世。然而,对于某些特定靶点,这些模型的预测性能显然仍显不足。为此,为提升LD50等靶点的预测效果,有研究者提出了一种共识模型,该模型整合了由35家不同机构构建的多种预测模型。此外,李等人还提出了一种新方法,即利用化合物的多重特征(指纹与MDs)、多种算法(支持向量机、随机森林、k近邻法及XGBoost)以及不同类型的评价指标(回归、多分类和二分类),共同构建预测模型。不过,上述方法往往需要针对单一评估目标分别选用35种或更多不同的预测模型,并且还需根据具体评估任务逐一挑选描述符和算法,这无疑耗费了大量精力来建立模型。因此,当面临多个预测目标且需随着新数据的不断积累而频繁更新模型时,这些方法在药物发现阶段的实际应用难度较大。由此可见,亟需开发出易于适用于多种评估任务、且能有效提升预测性能的新方法。
近期开发了一种名为DeepSnap-DL的深度学习方法,该方法通过将化合物的图像作为特征输入,构建高效的预测模型。研究表明,相较于传统机器学习方法,这一创新方法在预测药物靶点的毒性参数方面表现出更优异的性能,例如对组成型雄烷受体和芳烃受体的活性、线粒体膜电位以及清除率(CL)等指标的预测效果显著优于后者。尤其值得一提的是,在这些毒性参数中,研究团队还提出了一种创新策略,即将DeepSnap-DL方法与基于分子描述符(MD)的传统方法相结合,进一步提升了CL分类模型的预测精度。不过,目前这种方法的应用仍局限于CL参数的分析领域。
基于此,本研究旨在探索如何利用DeepSnap-DL方法与MD-based方法相结合,进一步提升各类分类模型的预测性能,以满足药物发现阶段的多样化评估需求。为此,我们选取了LD50作为毒性评估指标,并以血脑屏障穿透性(BBBP)和CL通路作为药代动力学评估指标,所用数据均来自公开数据库。其中,LD50定义为能使50%实验动物死亡的化合物剂量;由于作用于中枢神经系统的药物必须能够透过血脑屏障,因此其BBBP值通常要求较高。另一方面,CL通路主要涉及药物的排泄途径,可分为肝代谢和肾消除两大类。在预测人体CL通路时,选择合适的预测方法至关重要。值得注意的是,这些关键参数均可通过动物实验进行验证,但此类实验成本高昂且耗时较长,难以快速获得实验结果。因此,在化合物合成前,借助QSAR分析提前预测这些参数显得尤为必要。基于以上背景,本研究将在药物发现过程中,首次尝试将DeepSnap-DL方法与MD-based方法有机结合,以期显著提升上述评估指标的预测性能。
方法
通过综合应用多种数据处理和算法,作者构建了用于预测 LD50、BBBP 和 CL 途径的高效模型。
数据集的筛选和处理
为了构建 LD50 的预测模型,作者选择了基于 Mansouri 等人报告的「毒性很强」的数据集。与此同时,BBBP 和 CL 途径的数据集也经过了严格的筛选和处理。所有数据都被有目的地排序,并在 4:1 的比例下随机分成训练和测试集。
计算分子描述符(MDs)
在化合物的结构数据中,含有抗衡离子和水分子的化合物已通过使用Molecular Operating Environment (MOE) 2019.01版(日本东京MOLSIS公司)处理去除杂质盐类后,从数据集中移除。随后,研究团队应用RDKit软件,基于先前报道中的方法(35),分别以MMFF力场为依据,对每种化合物的LD50、BBBP及CL通路相关三维(3D)结构进行了优化。此外,MOE、alvaDesc(2.0.2,意大利莱科Alvascience srl公司)以及ADMET Predictor(9.5.0.16,美国加利福尼亚州兰卡斯特SimulationsPlus公司)也被用于计算分子描述符(MDs)。值得注意的是,在生成MDs时,ADMET Predictor会自动剔除所有字符串类型的描述符。最终,共有6554个描述符被选中,以供后续分析使用。
数据集划分为训练集与测试集,并通过化学空间分析进行验证
由于先前报告中已指定了LD50的训练集和测试集,因此本研究沿用了同一份来自先前报告的数据集。**在采用分层随机抽样法后,BBBP和CL通路的化合物被随机划分为训练集与测试集,比例为4:1。**此外,我们还利用JMP Pro软件14.3.0版(SAS Institute Inc., 北卡罗来纳州卡里市)中的主成分分析(PCA),选取了11个分子参数,以探讨模型的应用域。(20)本研究中评估的参数包括:分子量、Slog P值(辛醇/水分配系数的对数值)、拓扑极性表面积、h_logD值(pH=7时的辛醇/水分配系数)、h_pKa值(pH=7时的酸度)、h_pKb值(pH=7时的碱度)、a_acc值(氢键受体原子数)、a_aro值(芳香族原子数)、a_don值(氢键供体原子数)、b_ar值(芳香键数)以及b_rotN值(可旋转键数)。最终,我们计算出了三个主成分(PC1~PC3)。
DeepSnap
本研究使用了Java查看器软件Jmol,将三维化学结构以不同颜色的3D球棍模型呈现出来(图S1)。(14,16,17,36−39)在本研究中,这些三维化学结构被自动捕捉为三个轴(x轴、y轴和z轴)各145°视角下的快照。此外,用于DeepSnap展示过程的其他参数如下:图像像素尺寸为256×256(RGB),每SDF文件包含100个分子,缩放因子设置为100%,原子大小设定为范德华半径的23%,键半径为15 mÅ,最小键距为0.4 Å,键容差为0.8。值得注意的是,在根据目标变量对训练数据集进行排序后,这些数据集被随机划分为DeepSnap(训练)集与DeepSnap(验证)集,其比例为3:1,分别适用于BBBP、LD50及CL通路的研究。而DeepSnap-DL的数据集则包括DeepSnap(训练)、DeepSnap(验证)以及测试集三部分(图S2)。
DeepLearning
DeepSnap生成的二维图像快照被保存为PNG文件,并通过NVIDIA DL GPU训练系统(DIGITS)6.0.0版本软件,在配备四块Tesla-V100显卡(每块32 GB)的系统上进行了尺寸调整。我们采用预先训练好的深度学习模型Caffe,在Ubuntu 16.04LTS操作系统上,快速完成高精度卷积神经网络(CNN)的训练与微调。其中,深度CNN架构采用了GoogLeNet,优化则使用了Adam算法。在DeepSnap-DL方法中,预测模型基于DeepSnap训练数据集构建,训练过程涵盖15至300个Epoch,每个Epoch设置一个快照间隔和一个验证间隔;同时,每次训练均采用单一随机种子,学习率范围为10⁻⁷至10⁻³,批量大小、批处理累积策略、学习率调度策略、步长及Gamma参数均默认启用。DeepSnap验证数据集中的最低损失值,即为DeepSnap验证数据集与对应标注数据集结果的误差率,这一条件被用于测试集预测的评估。此外,利用DeepSnap-DL方法,针对以不同角度(X、Y、Z轴)捕捉的单个分子图像,分别计算出具有最低损失值(DeepSnap验证)条件下的概率。最终,所有预测值的中位数被用作目标分子的代表性数值。为了构建包含随机种子值为2至4的预测模型,研究团队还依据随机种子值为1时所确定的学习率与Epoch数,进一步完成了模型的搭建。
基于MD构建预测模型
模型构建与分析工作于2022年5月20日至2022年11月2日期间,采用DataRobot(SaaS,DataRobot,日本东京)平台进行。如前所述,DataRobot自动开展了包含多种算法选择及数据预处理技术的建模竞赛。(41,42)研究中实施了五折交叉验证,其他实验条件则按先前描述执行。(15)在依据内部验证的Logloss评分筛选出最佳模型后,我们利用排列重要性方法,从6554个候选MD中遴选出100个关键MD。随后,根据验证结果最终确定了14种建模算法,并构建了共计42个模型(表S7)。在完成内部验证的Logloss评分计算后,我们使用全部训练数据构建了表现最优的模型。该最终模型被用于评估测试集的预测性能(图2)。
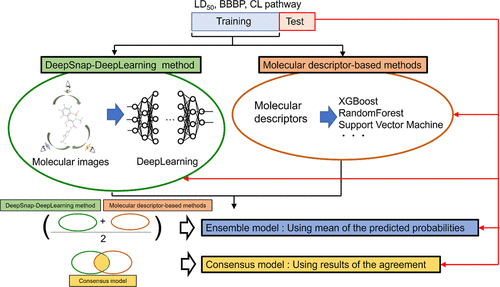
图2. LD50、BBBP及CL通路建模流程图。训练数据集被用于构建预测模型,分别采用DataRobot的基于MD的方法和DeepSnap-DL方法。随后,通过整合MD方法与DeepSnap-DL方法,建立了集成模型和共识模型。各预测模型的评估指标均基于测试集计算得出。LD50,半数致死剂量;BBBP,血脑屏障穿透性;CL,清除率;DL,深度学习。
本研究探讨了DeepSnap-DL方法与基于MD的方法的联合应用。在第一种方法中,将两种方法获得的预测概率进行平均,并将该值作为新预测模型(集成模型,图2)的预测概率;而在第二种方法中,则采用两种方法一致的结果(共识模型,图2)。
模型综合与评估
通过 DataRobot进行模型构建和分析,作者实施了五折交叉验证,并根据内部验证的 logloss 分数选定了最终的算法。作者还研究了深度学习和基于 MD 的方法的综合应用,包括平均预测概率和达成共识的模型。
通过应用多种评估指标如 AUC、ACC、BAC 等,作者精细地评估了每个模型在预测 LD50、BBBP 和 CL 途径方面的性能。最优的截断值是通过应用 Youden 指数来确定的。
结果
通过集成和共识模型,DeepSnap-DL 和 MD-based 方法能有效提高化学物质预测的准确度。
数据分离和化学空间分析
为了确认化合物分离的准确性,作者使用主成分分析(PCA)对 LD50、BBBP 和 CL 路径的数据集进行了验证。PCA 能有效揭示应用领域,这一点已由之前的研究证明。不同数据集在主成分的方差上表现出一致性,从而有效地将化合物分离为训练和测试数据集。
DeepSnap-DL 模型构建
在药物发现的筛选中,DeepSnap-DL 方法从多个角度进行了研究。本研究使用了五个学习率和五个最大迭代次数来构建模型。最终模型的选择基于曲线下面积(AUC)的最高值,从而确保预测的精准性。
MD-based 方法和模型
根据内部验证的 logloss 结果,作者从 6554 个 MDs 中选出了 100 个,用于构建最终的预测模型。不同的算法被选为每个模型的基础,而测试数据集的评估指标进一步证实了这一方法的有效性。
集成和共识模型的优势
集成模型通过平均 DeepSnap-DL 和 MD-based 方法的预测概率,达到了比单一方法更高的 AUC 和 BAC。而共识模型在去除不一致化合物后,虽然可评估化合物数量有所减少,但在评估指标上表现得更好。
图表解读
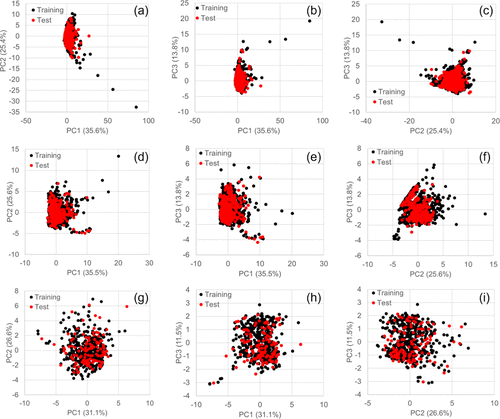
图 1: 展示了 11 个具代表性的基于 MD(Molecular Dynamics)的三组分主成分分析(PCA)得分图,用于 LD50、BBBP 和 CL 路径预测。
(a, d, g): PC1 和 PC2 的得分图。横轴和纵轴分别代表 PC1 和 PC2。
表 2: LD50、BBBP 和 CL 路径的外部测试结果。
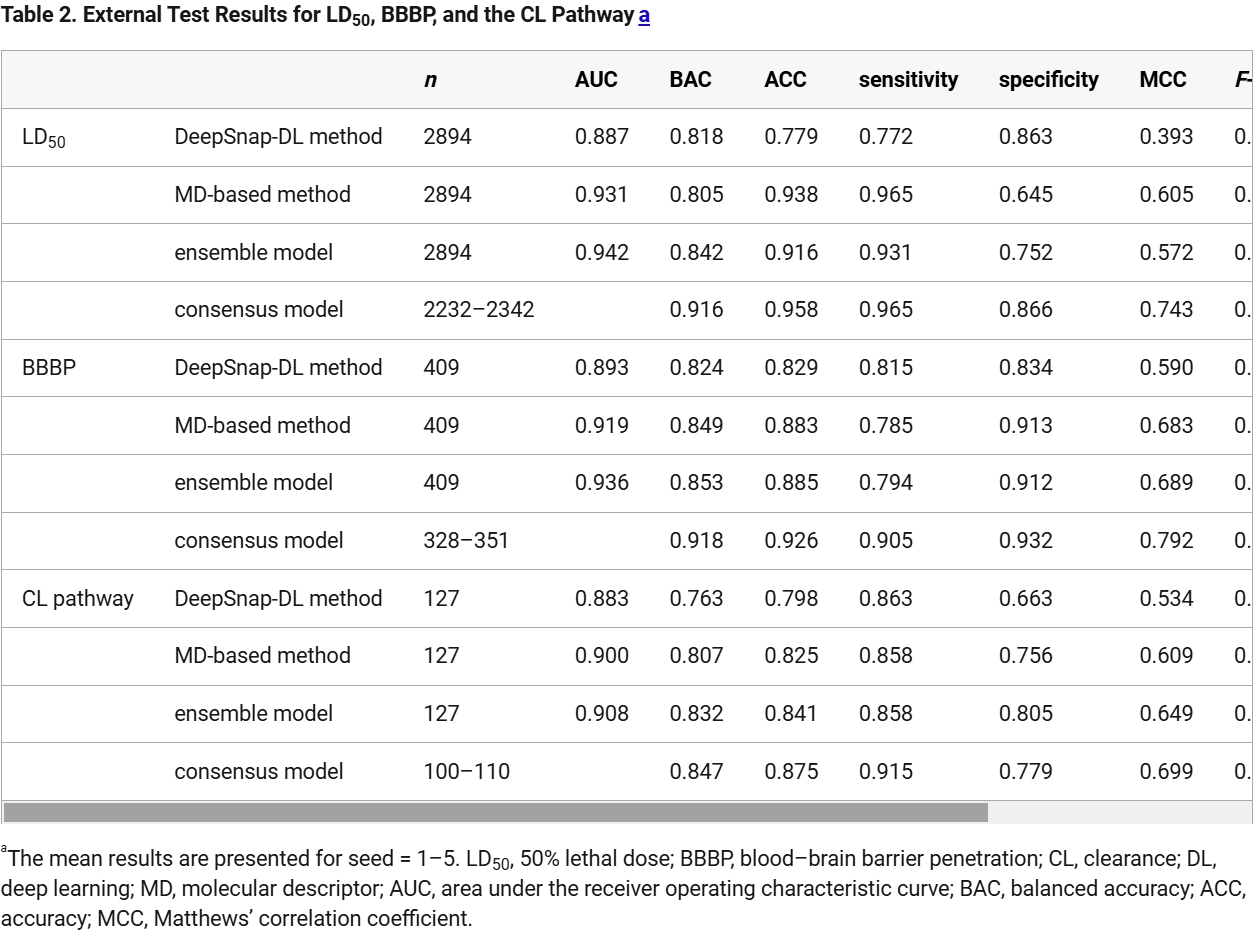
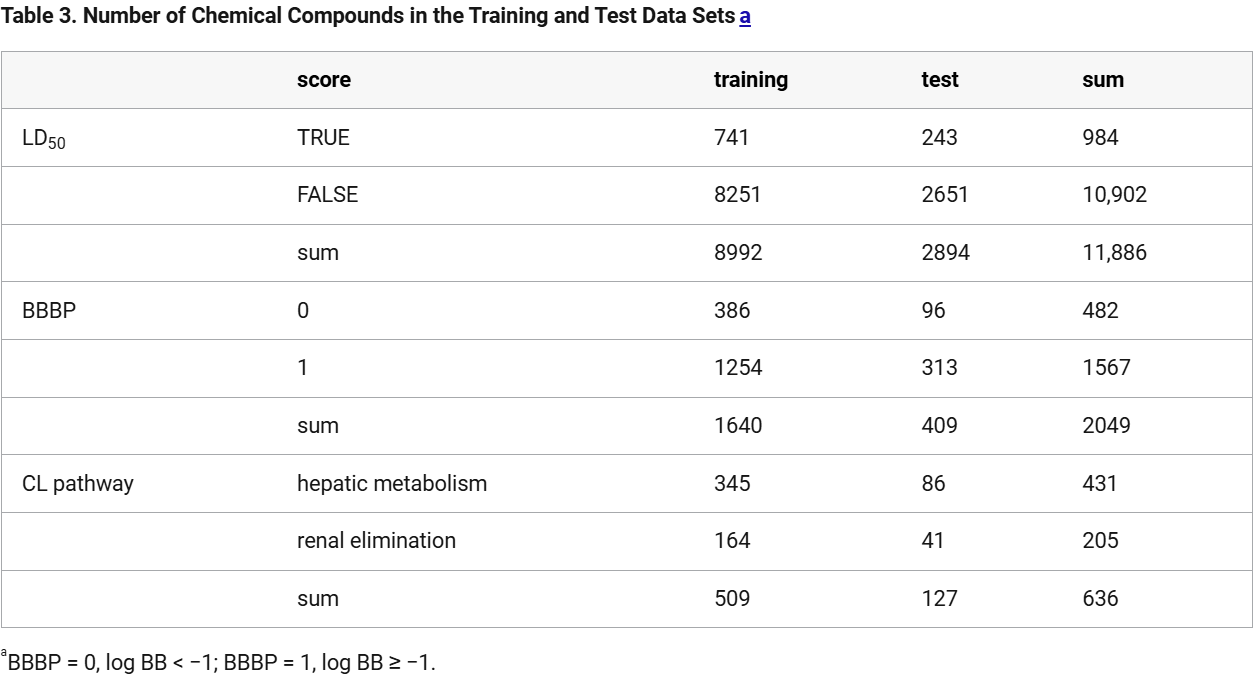
表 3: 训练和测试数据集中的化学化合物数量。
创新点
本研究通过结合 DeepSnap-DL 和 MD-based 方法,显著提升了 QSAR 模型在药物开发中的预测性能。
- QSAR 分析在减少动物实验和成本方面的重要性
QSAR(定量结构活性关系)分析已广泛应用于药物开发的早期阶段,用于预测药物的药理活性、药代动力学参数和毒性参数。这种方法依赖于分子描述符(MDs)和算法,如支持向量机、随机森林和深度学习等。因此,多家制药公司使用基于 QSAR 分析的预测模型进行虚拟筛选和实验优先级的确定。 - 本方法与前人工作的比较
尽管前人的研究已经报道了多种基于 QSAR 分析的预测模型,但其中一些模型在某些目标上的预测性能显然不足。与之不同,本研究采用了结合 DeepSnap-DL 和 MD-based 方法的新模型,不仅提升了毒性参数的预测性能,还增强了对药代动力学参数的准确度。 - 实验结果证明了新模型的优越性
在使用多种条件进行深入探究后,作者发现本研究的新模型在 LD50、BBBP 和 CL 通路方面都具有高度的预测性能。特别是,与 CATMoS 等前人工作相比,本模型在预测准确性和相关性方面都有明显的优势。
结论
通过结合 DeepSnap-DL 和 MD-based 方法,作者在 LD50、BBBP 和 CL 路径方面实现了预测性能的显著提升。
预测模型的优化
本研究集中在 DeepSnap-DL 和 MD-based 方法的结合,针对 LD50、BBBP 和 CL 路径进行预测。作者通过多样性的超参数调整,实现了预测性能的明显提升。具体来说,作者发现学习率和最大周期数对模型性能有显著影响。
动物实验的替代方案
动物实验是药物发现中不可或缺的一环,但因成本和伦理问题,近年来有多种替代方法在探索中。本研究使用了基于 QSAR 分析的预测模型,并与其他研究进行了比较,证明了其有效性和准确性。
中枢神经系统药物分布
对于作用于中枢神经系统的化合物,评估其在大脑内的分布至关重要。作者的模型在 BBBP 数据集上的表现优于其他多数模型,证明了其在替代动物实验方面的潜力。
数据集与性能
本研究使用了多个不同的数据集,并通过精心设计的实验,证明了预测模型的有效性。尤其是在 CL 路径预测方面,作者的模型性能超过了先前研究。
实用性与局限性
虽然共识模型表现最佳,但它不能评估所有测试化合物。因此,在药物筛选的早期阶段,首先使用共识模型进行评估,然后对不能被评估的化合物使用集成模型,可能是一种实用的方法。
通过本研究,作者证明了通过结合两种预测模型,能够简单而有效地构建高性能的预测模型。这种方法不仅加速了药物发现过程,还为未来研究提供了新的方向。
Mamada H, Takahashi M, Ogino M, Nomura Y, Uesawa Y. Predictive Models Based on Molecular Images and Molecular Descriptors for Drug Screening. ACS Omega. 2023 Sep 13;8(40):37186-37195. doi: 10.1021/acsomega.3c04073. PMID: 37841172; PMCID: PMC10568689.