【AI4S】3DSMILES-GPT:基于词元化语言模型的3D分子生成
3DSMILES-GPT:基于词元化语言模型的3D分子生成
- 引言
- 方法
- 骨干
- 可拆卸口袋编码器
- 强化学习
- 数据集与数据预处理
- 基准
- 训练与生成协议
- 结果
- 生成构象的质量
- 分子特性与结合模式
- 基于结构的药物设计,针对特定靶点
- 结论
- 数据可用性
分子生成是药物先导物发现和优化领域的一项重要创新技术,但当前这类方法在生成分子的有效性、结构合理性和合成可行性等多属性优化方面存在挑战,精度和效率往往难以两全。 近日,浙江大学药学院康玉副教授、侯廷军教授和谢昌谕教授以及华为刘力维研究员团队在分子生成领域取得突破,成功研发了一种全新的基于纯语言模型的分子生成框架:3DSMILES-GPT。这一框架通过将分子的二维和三维结构视为语言表达,在语言模型的帮助下,实现了药物分子的高效生成。 相关成果以 “3DSMILES-GPT: 3D Molecular Pocket-based Generation with Token-only Large Language Model为题发表在Chemical Science上,并入选为2024 Chemical Science HOT Article Collection。

- 3DSMILES-GPT: 3D molecular pocket-based generation with token-only large language model
- Jike Wang,‡ Hao Luo,‡ Rui Qin,‡ Mingyang Wang, Xiaozhe Wan, Meijing Fang, Odin Zhang, Qiaolin Gou, Qun Su, Chao Shen, Ziyi You, Liwei Liu,* Chang-Yu Hsieh,* Tingjun Hou* and Yu Kang*
- Chem. Sci., 2025,16, 637-648
- https://doi.org/10.1039/D4SC06864E
基于目标结构生成三维(3D)分子,是药物发现领域的一项前沿挑战。然而,目前许多现有方法往往会产生构型无效、不符合物理实际、药效不佳、合成难度大,且耗时较长的分子。为应对这些难题,我们提出了3DSMILES-GPT——一种完全由语言模型驱动的3D分子生成框架,该框架仅使用标记符进行操作。我们将二维(2D)和三维分子表示均视为语言表达形式,通过全维表示将其有机结合,并在包含数千万种类似药物分子的海量数据集上对模型进行预训练。这种纯标记符的方法使模型能够全面理解大规模分子的二维与三维特性。随后,我们利用蛋白质口袋与分子的成对结构数据对模型进行微调,并进一步采用强化学习技术,以优化所生成分子的生物物理及化学性质。实验结果表明,3DSMILES-GPT生成的分子在结合亲和力、药物相似性(QED)以及合成可及性评分(SAS)等方面,均显著优于现有方法。尤为突出的是,其在QED定量评估方面实现了33%的性能提升,同时通过Vina对接估算的结合亲和力仍保持行业领先水平。此外,该模型的生成速度极为高效,平均每生成一个分子仅需约0.45秒,较现有最快方法提升了三倍。这一创新的3DSMILES-GPT方法,有望为药物发现领域中3D分子的高效生成带来积极影响。
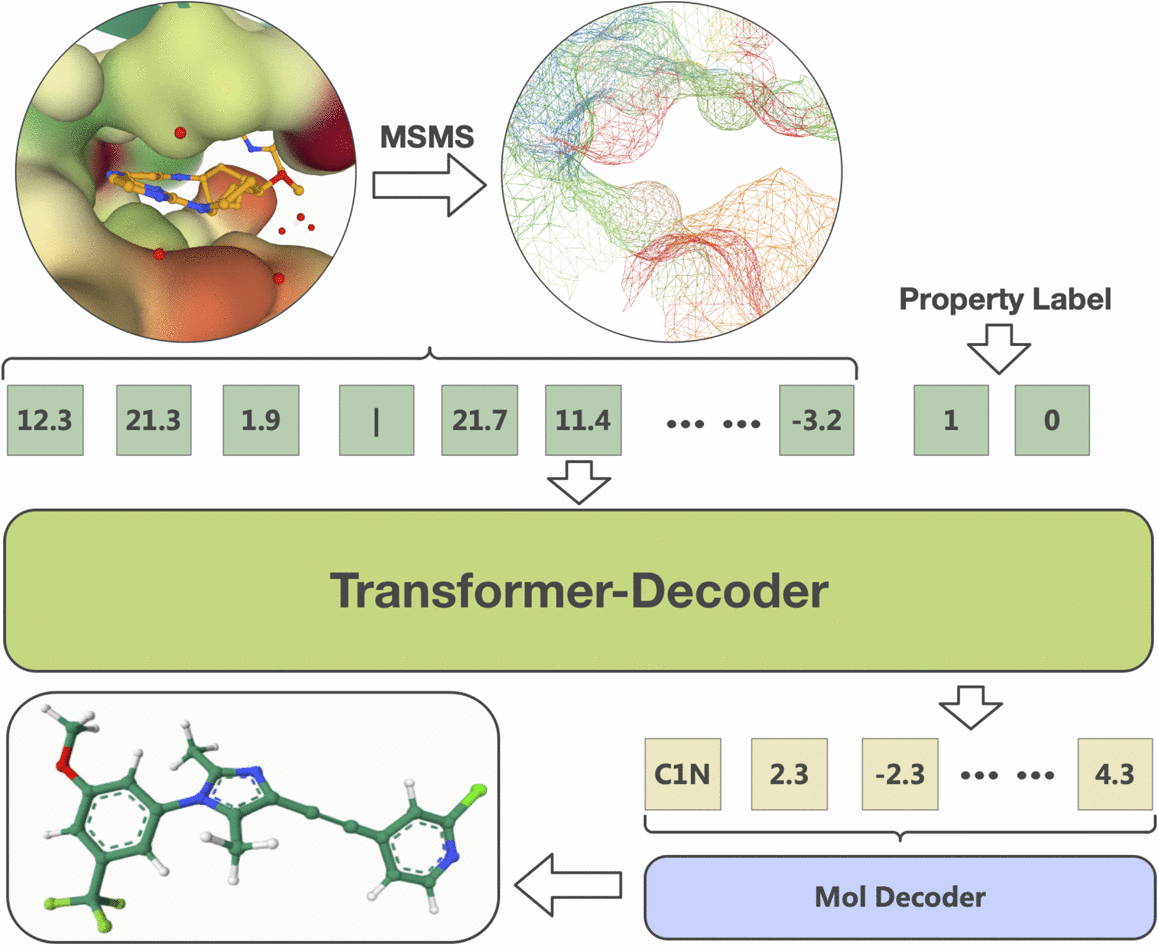
引言
近年来,深度生成模型备受关注,在自然语言处理到视频合成等多个领域均展现出令人瞩目的进展。这些模型在连续型数据域中的编码与合成能力尤为出色。然而,随着研究重心逐渐转向更为复杂且离散的数据类型——尤其是化学分子——人们愈发重视开发能够生成真实可靠、高效实用分子数据的生成模型。正是在这一背景下,深度生成模型的不断进步,推动了多种相关方法学的兴起,旨在攻克分子生成这一难题,为创新性药物分子设计开辟了广阔前景。
在过去一段时间里,基于配体的分子生成(LBMG)技术广受欢迎。根据生成分子的具体表示方式,这类方法主要可分为两大类:基于图的分子生成和基于序列的分子生成。其核心原理是将分子分别以图或序列形式进行表征,从而将分子生成任务转化为图结构生成或自然语言生成问题。此外,研究人员还借助贝叶斯优化(BO)和强化学习等技术,引导模型精准生成理想的药物分子。
分子本身天然具有类似图的结构,因此用图形方式表达其信息显得直观易懂。正因如此,基于图表示和传统启发式算法的分子设计方法早已成熟。例如,布朗等人于2004年利用遗传算法,提出了一种以分子图为基础的优化算法;而2013年,维尔舒普等人则推出了ACSESS算法。近年来,随着图神经网络(GNN)的快速发展,这类网络在处理各种基于图结构数据的复杂问题时展现出非凡的适应能力。德考等人于2018年率先将GNN引入药物设计领域,提出了MolGAN模型,从而为分子设计开辟了全新路径。随着基于图的方法不断取得突破性进展,越来越多的研究人员开始借助分子图表示来进行药物设计。
与基于GNN的分子合成策略相比,基于序列的方法提供了一条更为简洁的途径。这主要得益于化学化合物能够通过诸如简化分子输入行输入系统(SMILES)或SELFES等化学语言得到有效表征,这些语言的结构甚至与自然语言极为相似。因此,众多关于分子设计的学术研究纷纷提出了基于循环神经网络(RNN)或Transformer架构的框架。2016年,ChemVAE将变分自编码器(VAE)与贝叶斯优化相结合,探索潜在空间,以寻找具备特定属性的分子。2017年,奥利克罗纳等人则利用强化学习对基于RNN的分子生成过程进行精细调优,成功生成出与目标结构相似或具备预定活性的分子。2021年,王等人改用Transformer解码器替代RNN进行分子生成,并结合知识蒸馏与强化学习,开发出了MCMG模型。随着时间推移,多种基于序列的分子生成方法相继涌现。
然而,二维分子生成方法存在一个显著的局限性:这些技术忽略了蛋白质口袋与分子之间至关重要的三维结构互补性。鉴于配体-蛋白构象选择在药物设计中的核心作用,要准确评估这种互补特性,必须基于蛋白质口袋和分子本身的内在三维结构来深入理解。因此,近年来,基于三维结构的分子生成方法逐渐受到广泛关注。
随着深度几何学习的兴起,一系列关于自回归式三维分子生成的研究相继涌现。例如,Gebauer提出了G-SchNet模型——这是一种自回归的深度神经网络,能够通过在欧氏空间中依次定位原子,高效生成多样化的有机小分子。随后,诸如LiGAN、GraphBP、SBDD以及Pocket2Mol等模型也被开发出来,直接实现在蛋白质口袋内生成目标分子。尽管如此,自回归方法容易出现误差累积的问题,这促使研究者们进一步探索基于扩散的三维分子生成新途径。这类方法无需逐个生成原子,而是能一次性完成整个分子的构建。不过,截至目前,这些策略仍未能有效捕捉化学键的分布特征,导致生成的分子结构往往不具实际应用价值。
上述讨论的3D分子生成方法主要依赖于图神经网络(GNN)。尽管语言模型(LM)在2D分子设计过程中能够从海量类药化合物数据集中高效提取丰富的2D分子信息,但其对连续3D分子架构的表征能力仍显不足。然而,随着大规模语言模型(LLM)的迅速普及,已有大量研究表明,LLM可巧妙地学习到连续的数值表示。Born等人提出了回归Transformer模型,该模型通过将数值编码为标记,实现了统一的回归与预测任务。这一方法展示了Transformer在回归任务中的潜力——即将数值信息转化为标记进行处理,但其在捕捉复杂3D分子结构方面仍存在局限性,且当扩展至更大规模数据集时,效率可能受到影响。此外,Flam-Shepherd等人则采用笛卡尔坐标xyz标记来表示分子的3D结构,不过,由于标记化的离散特性,这种方法在生成符合物理规律的构象时可能面临挑战,并可能导致化学有效性的难以维持。尽管如此,这两种方法在其各自的药物设计应用中均已展现出可观的效果。最近,Feng等人提出了一种基于片段、以语言模型为核心的3D分子生成模型——Lingo3DMol,经基准测试评估,该模型的表现显著优于基于图网络的方法,展现出巨大潜力。
上述研究充分表明,语言模型在揭示分子固有3D结构特征的精细细节方面具有独特优势。相较于复杂的扩散模型和基于GNN的分子生成方法,依托语言模型的自回归方法不仅训练过程更为简单高效,而且仅依赖标记的范式还能无缝对接现有的通用语言模型。因此,我们积极探索一种更简洁、更直观的方法,用于精准刻画分子及蛋白质口袋的结构特征。在此基础上,我们证实大型语言模型(LLM)能够有效捕捉并利用与分子和蛋白质口袋相关的位置信息,从而实现针对目标口袋内新分子结构的高效生成。为此,我们推出了3DSMILES-GPT——一个创新的纯标记框架,专为明确的3D分子生成而设计,完全基于大型语言模型构建。如图1所示,3DSMILES-GPT的核心架构是一套Transformer解码器。通过将2D和3D结构的生成任务视为自然语言生成问题,3DSMILES-GPT将原子的3D坐标编码为标记,进而高效获取分子的二维和三维信息。为了充分发挥语言模型的内在能力,我们的方法首先利用包含大量类药分子的数据集对3DSMILES-GPT进行预训练;随后,在特定的蛋白-配体数据集上进行微调,并结合口袋表面的原子坐标以及配体分子信息。此外,为增强模型从蛋白质口袋中提取信息的能力,我们还引入了一个可拆卸的模块化蛋白质编码器。同时,借助强化学习方法,我们进一步优化了生成分子在多种关键属性上的表现。实验结果表明,与当前最先进的(SOTA)方法相比,3DSMILES-GPT在8项基准指标中均取得了最优性能,涵盖生物活性、类药性及合成可及性等多个维度。此外,针对5个不同靶点蛋白开展的专项案例研究也进一步验证了其在实际场景中生成具有强结合力的类药分子方面的卓越能力。
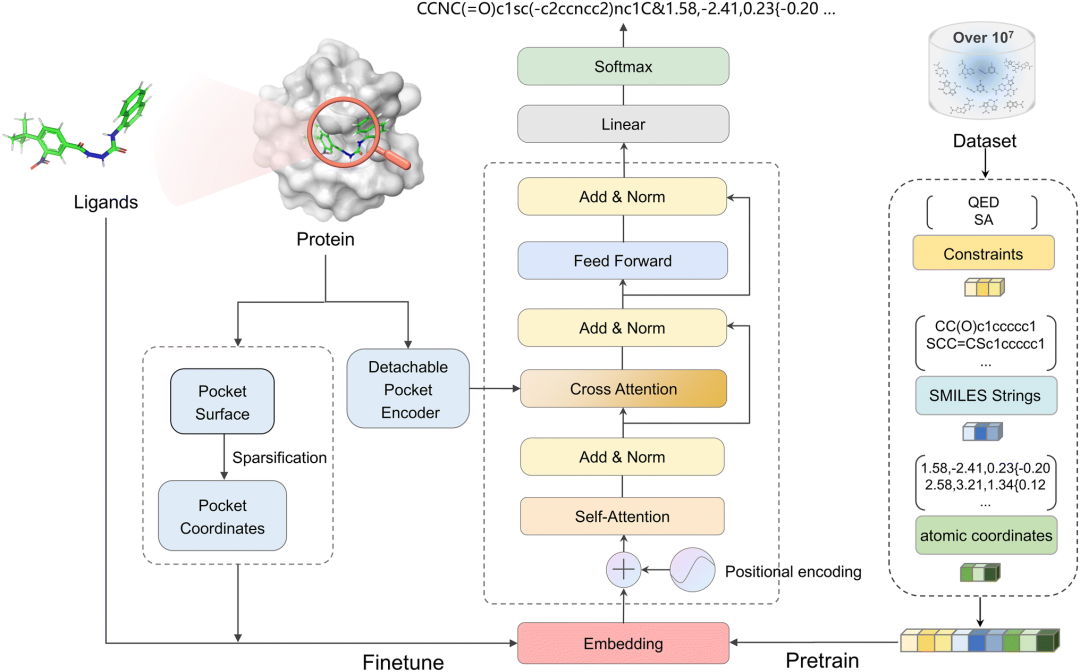
图 1. 3DSMILES-GPT 模型架构。3DSMILES-GPT 方法通过在大型药物样分子数据集上预训练模型,使其能够在保证结构合理性的前提下,快速生成具有良好成药性的分子。模型以 Transformer 解码器为骨架,通过将生成二维和三维结构的任务构建为自然语言生成的问题,将原子 2D 结构和 3D 坐标编码为字符,从而有效捕获分子的 2D 和 3D 信息。模型首先在 PubChem 数据集上进行预训练,然后在特定蛋白质-配体数据集上进行微调。此外,为了更好地提取蛋白质口袋信息,3DSMILES-GPT 引入了一个可拆卸蛋白质编码器。
方法
骨干
3DSMILES-GPT 的架构包含一个具有 12 个注意力头的 8 层 Transformer 解码器,能够高效地实现二维和三维分子结构的自回归预测,并以直观的方式加以表达。多头注意力机制是 Transformer 模型的核心组成部分,使模型能同时关注输入数据的不同子空间,从而捕捉更丰富的信息。在多头注意力机制中,每个注意力头会学习一组权重,用于计算输入序列中不同位置的注意力权重,这些权重随后被用来加权输入序列的表示。通过在多个注意力头之间并行开展计算,模型得以从多种视角解读输入序列,进而提升其表征能力和泛化性能。注意力机制的具体形式如式 (1) 所示:
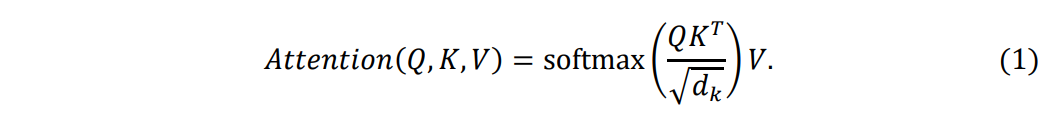 其中,Q、K和V分别表示查询矩阵、键矩阵和值矩阵,而dkd_kdk是K的维度。
其中,Q、K和V分别表示查询矩阵、键矩阵和值矩阵,而dkd_kdk是K的维度。
可拆卸口袋编码器
为增强蛋白质口袋信息的提取,我们设计了一种可拆卸口袋编码器。我们采用了Zhou等人提出的空间位置编码策略,该策略基于高斯核函数来描述原子间的相对位置。原子对之间的D维位置编码可由以下公式表示:
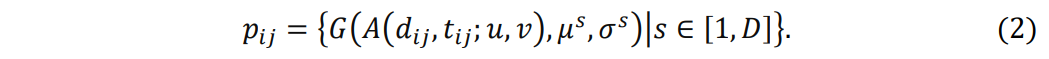 其中,G 表示高斯密度函数:
其中,G 表示高斯密度函数:
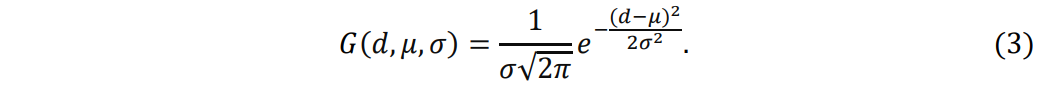
A 表示以uuu、vvv 参数化的仿射变换:
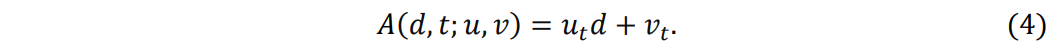
因此,iii 和 jjj 的信息可计算如下:
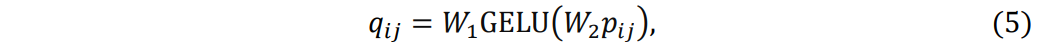
其中,W1W_1W1和W2W_2W2是可学习的参数。最终,通过交叉注意力机制,实现了口袋编码器与主干网络之间的融合。
强化学习
利用强化学习来提升基于序列的分子生成模型(如REINVENT),已是一项成熟的技术。然而,将其应用于在蛋白质口袋中生成三维分子这一特定任务,目前仍相对少见。通过简化三维分子生成的复杂过程,并将分子坐标表示为标记,我们能够更便捷地直接采用类似REINVENT的方法,以优化生成的分子。不过,在此背景下,我们选择了一种更为明确的策略:即通过多次迭代,结合策略梯度技术,逐步精炼模型。
在当前情境中,Pθ(M∣C)P_θ(M|C)Pθ(M∣C) 表示我们模型所遵循的初始策略,其中CCC代表蛋白质口袋,MMM则指代分子;同时,D0D_0D0 为初始微调数据集。在每一轮迭代中,我们会针对每个蛋白质口袋采样KKK个分子,并将那些表现出优异特性的分子纳入第ttt次迭代的微调数据集DtD_tDt,以供后续迭代使用。在整个微调过程中,我们将持续运用策略梯度方法,对模型的策略Pθ(M∣C)P_θ(M|C)Pθ(M∣C) 进行逐次优化与精进。
数据集与数据预处理
预训练阶段从PubChem药物数据集中筛选出1000万种分子,其中排除了原子数超过48或含有“C”、“O”、“N”、“S”、“P”、“F”、“Cl”、“Br”和“I”以外元素的分子**。每种分子均利用RDKit进行立体异构体枚举,随后为每个立体异构体生成两种构象,并采用MMFF94力场对这些构象进行能量最小化处理。构象中心化步骤则是通过从每个构象中减去其坐标中心来实现的。
在微调阶段,我们采用了CrossDocked2020数据集,并遵循Pocket2mol方法学,同时剔除了RMSD大于2 Å的对接姿态。以配体为中心,围绕其6 Å范围内的残基区域被划定为口袋数据;接着,利用MSMS工具计算出口袋表面的坐标,并通过pymesh对这些坐标进行了稀疏化处理。
此外,我们还对数据进行了进一步处理,以满足模型的输入需求。首先,计算了分子的QED值和log P(分配系数)值。其中,QED值高于0.5或log P值介于-1至3之间的分子被标记为1,而超出此范围的分子则标记为0。在微调阶段,我们同样采用类似的方法对训练数据进行标记,并额外加入了Vina评分标签:Vina评分低于-0.75的分子被标记为1,而评分等于或高于-0.75的分子则被标记为0。
对于二维分子结构的表示,我们使用了SMILES编码方式,并将SMILES序列以字符字节级别而非字节级标记化的方式进行编码。初始词汇表由从SMILES字母表中提取的72个字符组成,经过标记化后,最终被划分为1000个最常出现的标记。
针对三维分子结构,我们采用了数据增强技术,以赋予模型三维等变性特征。具体而言,通过对三维结构进行随机平移和旋转操作实现数据增强,其中每个坐标都被单独表示为一个独特的标记。在数据增强的具体细节上,每种分子都会围绕X、Y和Z轴进行随机旋转,且各轴的旋转角度独立地从0°到360°均匀采样。这种方法确保模型能够接触到分子的所有可能方向,从而有效促进旋转不变性。同时,我们还对分子坐标施加随机平移,平移向量则在-10 Å至10 Å的范围内均匀采样,以引入细微的位置变化,但又避免大幅偏离分子的原始位置。为了保证增强后数据集的一致性,所有分子均统一应用相同的增强技术,从而维持原有分子特性的分布状态。
基准
我们选择了代表三种不同口袋生成方法的SOTA模型:基于GNN的自回归模型Pocket2Mol;采用扩散方法实现一次性生成的TargetDiff;以及源自语言模型的Lingo3DMol。其中,Pocket2Mol和TargetDiff均使用CrossDocked2020数据集进行训练,而Lingo3DMol则首先在包含1200万种类药物分子的数据集上进行了预训练,随后在PDBbind2020 55数据集上进行了微调。在本研究的评估中,我们直接采用了各相关研究提供的代码及预训练模型。
训练与生成协议
在训练阶段,我们首先将分子的QED和logPlog PlogP标签作为前缀添加到SMILES字符串中,随后将相应原子的坐标追加到SMILES字符串的尾部。对于相同原子之间的坐标,用逗号分隔;而不同原子间的坐标则用大括号括起来。
在微调阶段,我们将经过处理的蛋白质口袋表面坐标转换为前缀形式的输入字符串,使模型能够理解配体坐标的边界。序列的起始和结束分别由‘〈s〉’和‘〈/s〉’标记表示。
在整个训练过程中,我们采用自监督学习方法,帮助模型熟悉分子的SMILES字符串和坐标字符串。主要的优化目标是通过最小化负对数似然来实现,具体如公式(6)所示:
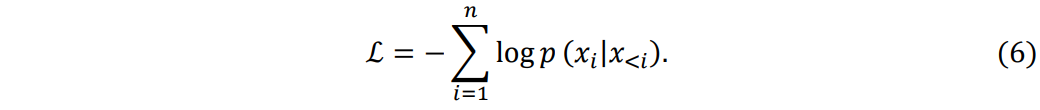
这一目标是通过不断迭代优化损失函数,利用梯度下降法直至收敛来实现的。
在生成阶段,我们将处理后的蛋白质口袋信息与指定的分子属性相结合,构成了模型的输入。用于生成SMILES和坐标字符串的自回归过程遵循公式(7):
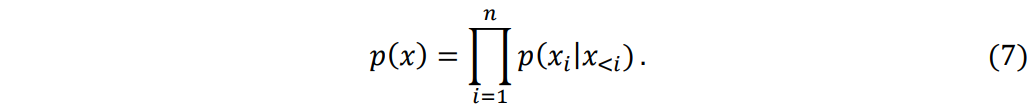
结果
语言模型在生成二维分子构型方面的能力毋庸置疑,这得益于它们对离散数据的出色处理能力。通过使用SMILES等化学语言作为输入,这些模型能够高效地掌握分子固有的二维拓扑排列特征。然而,一个关键问题仍待解决:语言模型能否有效捕捉连续数据的分布,包括分子构象?因此,在本部分中,我们首先评估3DSMILES-GPT生成的构象质量,随后分析所生成分子的性质及其结合效力,最后探讨3DSMILES-GPT针对特定药物靶标的泛化能力。
生成构象的质量
基于深度学习的分子生成面临一个重大挑战:频繁产生不符合物理实际的结构,而这一问题在现有的纯标记语言模型方法中尤为突出。这种局限性源于纯标记语言模型在有效处理连续数据时固有的困难,往往导致非物理可行的构象被生成。为解决这一关键问题,我们旨在识别那些能够展现真实物理构象的分子。在此基础上,我们将深入探索如何提升分子的结合亲和力,并优化其类药特性,同时将蛋白质口袋作为重要约束条件纳入考量。与单纯生成构象不同,本评估环节的核心聚焦于所生成分子在蛋白质口袋内的物理合理性。
对于结合在蛋白质口袋内的分子,由于柔性降低导致自由度受限,每种化学键的键长都呈现出稳定的分布特征,且不同受限口袋之间的键长差异并不显著。为评估模型性能,我们基于多种化学键类型,采用Jensen-Shannon散度(JSD)作为量化指标,比较了生成分子与训练分子中部分常见化学键长的分布情况。根据化学键类型对数据进行分类后,我们直观地展示了各键长的分布特征(图2a)。结果显示,我们的方法在整个化学键类型上均保持了最为均衡的表现,未出现明显短板——所有化学键类型的JSD值均未低于0.4。特别值得注意的是,在图2a前两个展示的类别中,即包含构成药物分子基本骨架的碳原子所形成的键,3DSMILES-GPT的整体表现略显不足,略逊于基于扩散模型的SOTA方法TargetDiff。不过,令人鼓舞的是,针对大多数与卤族元素相关的化学键,我们的模型反而取得了最佳效果。此外,当考察其他类型的化学键时,我们发现以JSD衡量的键长质量普遍分布在0.4至0.6之间,表明不同键型间的偏差较小,波动可控。尽管这些键在药物分子中的出现频率相对较低,但它们在硝基和磺酸基等关键功能基团中却至关重要,而这类基团常存在于抗菌药物中。由此可见,3DSMILES-GPT在这些键上的出色表现,很可能得益于GPT模型强大的信息保留与记忆能力,使其能够从稀少样本中精准复现含有这些罕见基团分子中原子的相对位置。
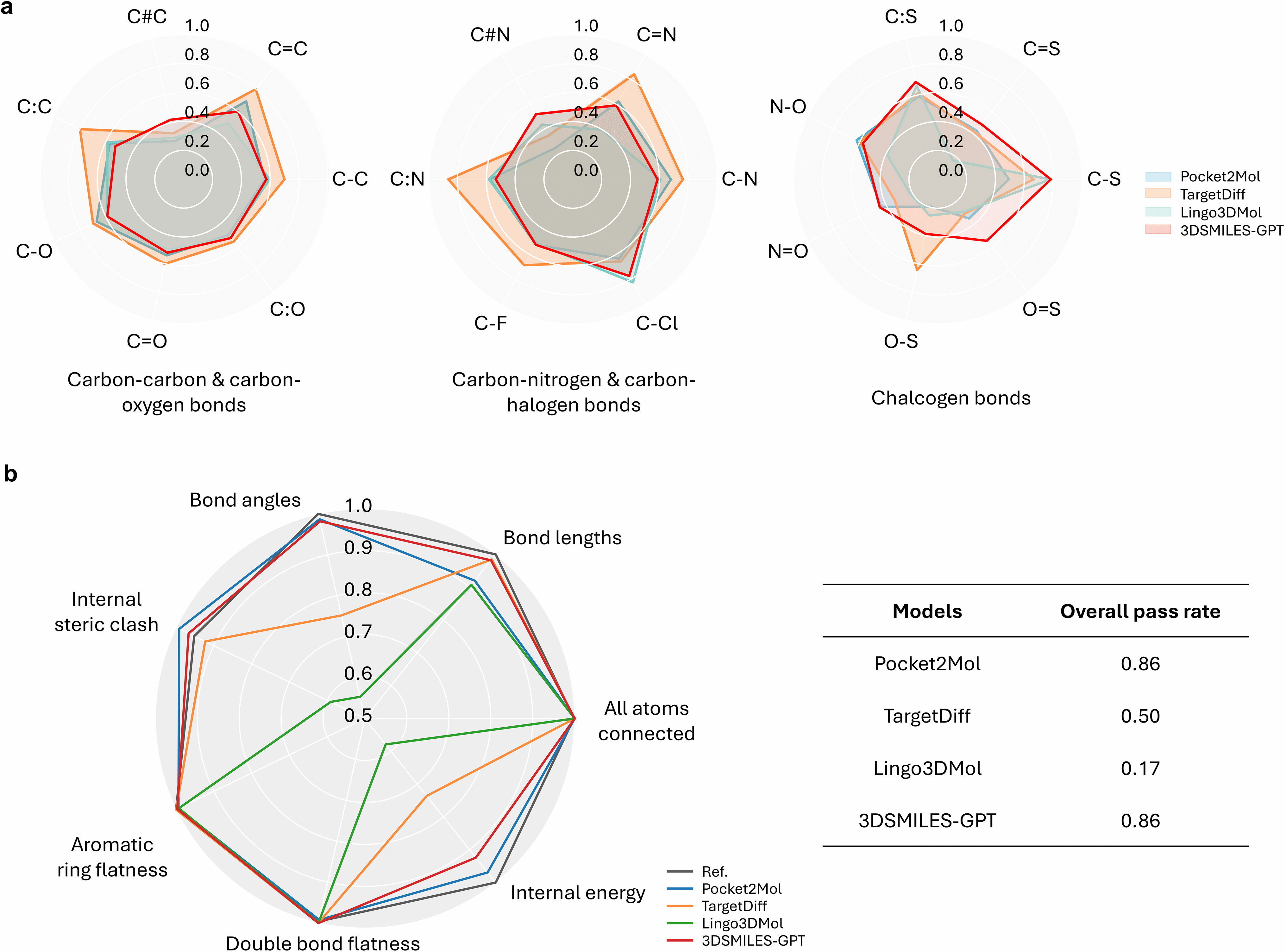
图2. 生成构象的质量。(a)3DSMILES-GPT与其他模型在常见化学键长上的Jensen-Shannon散度(JSD),并与参考分子进行对比。为便于直观比较,数值以1-JSD形式呈现,其中越接近1的值表明性能越佳。(b)各模型经PoseBusters测试后,在各项指标上的表现及其整体通过率。
如表S1†中更详细地所示,我们的模型在超过三分之一的键长类型上实现了最佳性能,而在其他键长类型中,其表现也与现有模型不相上下,甚至更为出色。在全球范围内,尽管我们的模型在口袋生成任务中的键长预测能力仍略逊于TargetDiff,但这种差距可能源于迁移学习过程中潜在的记忆遗忘现象。研究结果表明,与其他专为利用图神经网络(GNN)或语言模型进行口袋生成任务而设计的方法相比,我们模型的表现基本相当,甚至在某些情况下更为优异。这一发现进一步证实,以原子坐标作为预测的“标记”能够有效重现分子键的分布特征。
为进一步评估,我们采用了PoseBusters工具集——一套专门用于检测对接和分子生成过程中物理与化学一致性的软件工具。**该工具提供了多种指标,可全面检查分子构象中可能存在的误差。**因此,我们致力于确保所生成的分子在所有评估有效性、子结构及立体化学合理性的指标上均达到高通过率,而不仅仅是在个别特定指标上表现突出。从图2b和表S2†中可以看出,我们的模型在多个指标上的通过率始终稳定保持在85%以上,这表明绝大多数生成分子在物理和化学层面均符合自然状态下的合理性标准。相比之下,Lingo3DMol和TargetDiff等其他模型虽然在单个指标上表现出色,但在某些特定指标(如键角或空间位阻冲突)上却明显不足,其通过率仅介于50%至70%之间。而与Pocket2Mol相比,我们的模型在多个指标上的表现几乎不分伯仲,其中在各项独立指标上的通过率更是高达90%以上。此外,借助PoseBusters进行的键长分析,进一步验证了基于JSD的键长分析结果:3DSMILES-GPT和TargetDiff在键长指标上的通过率几乎达到100%,而其他模型的通过率则普遍低于80%。这充分说明,3DSMILES-GPT生成的大部分分子其键长均处于可接受范围内,与图2a中呈现的结果一致——即相对于参考分子,绝大多数化学键的JSD值均匀分布在理想的0.4至0.6区间内。
分子特性与结合模式
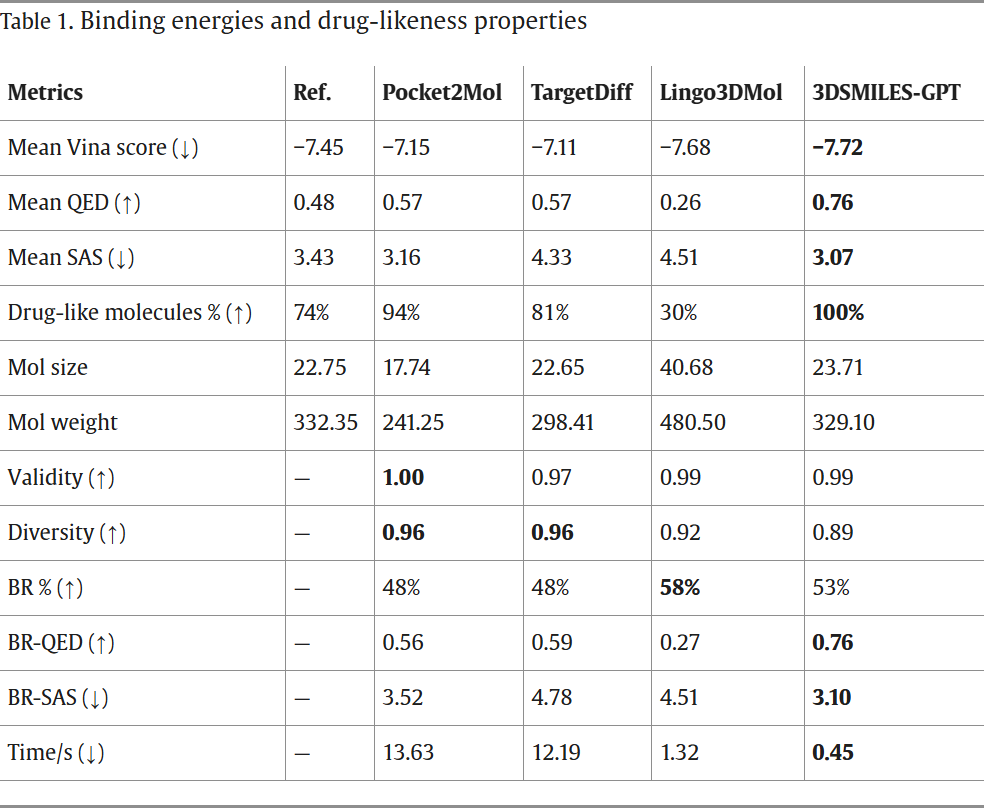
最初,通过QuickVina 2工具,直接利用Vina评分(kcal mol⁻¹)对生成分子的结合强度进行了评估。如表1所示,3DSMILES-GPT模型所生成分子的平均Vina评分显著高于其他基线方法,甚至超越了真实分子的表现。
通常,分子尺寸较大的结构更容易占据蛋白质的活性位点,从而带来更高的Vina评分。这一现象凸显了全面考量生成分子理化性质的重要性。正如Feng等人的研究指出,某些大型、多环结构的分子在药物研发中往往并不适用。因此,对生成分子进行深入评估至关重要。从表1的数据可以看出,与其它基线方法相比,我们模型生成的分子在分子量方面更接近真实药物分子;而在分子尺寸上,TargetDiff则与真实分子最为接近,表现出与我们的模型相似的特征。相比之下,Pocket2Mol和Lingo3Dmol生成的分子在尺寸和重量上分别偏小和偏大。
接下来,我们进一步分析了生成分子的SAS(合成可及性评分)和QED(类药性评分)指标,并将其与各基线方法进行了对比。结果显示,相较于其他基于口袋信息的分子生成方法,我们的模型在QED和SAS两项指标上均展现出明显优势。尤为突出的是,与表现最佳的基线方法Pocket2Mol相比,我们的模型在QED性能上实现了约33%的提升。这充分说明,3DSMILES-GPT生成的分子具有更为优异的类药特性,从而显著提升了其潜在的药物开发价值。此外,与其他基线模型相比,3DSMILES-GPT生成的分子还表现出更高的SAS值,表明其合成可行性更佳。同时,我们的方法在分子生成速度上也遥遥领先——经NVIDIA Tesla V100 GPU测试,每生成一个分子仅需0.45秒。
为进一步探究QED、SAS及其他多种分子特性之间的相互关系,我们采用了热图可视化技术(图3)。图3a展示了分子数量与其对应的QED和SAS值之间的关联。值得注意的是,3DSMILES-GPT生成的分子大多集中在左下角区域,这表明该模型生成的分子普遍具有较高的QED值和较低的SAS值,明显优于其他方法。此外,我们还考察了分子量对该关系的影响,发现Pocket2Mol生成的分子虽然QED较高、SAS较低,但其分子量却相对较小(图3b)。最后,我们进一步分析了Vina评分与QED/SAS之间的相关性。如图3c所示,3DSMILES-GPT生成的分子在左下角区域呈现出明显的浅色分布,这意味着其Vina评分较低,结合能也相应减弱。
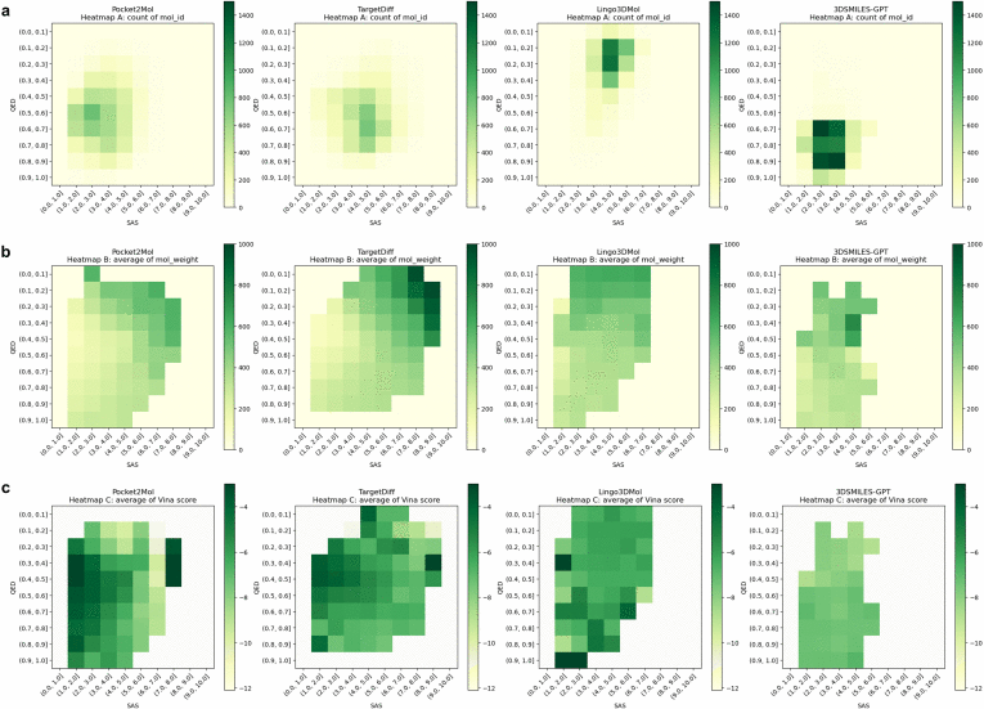
图3. 各模型生成分子的QED、SAS及其他性质的分布热图。(a)QED、SAS与分子数量,(b)QED、SAS与分子量,以及(c)QED、SAS与Vina评分。
在我们创造分子的探索过程中,我们的目标是开发出性能超越现有化合物的新分子。为了评估模型实现这一目标的有效性,我们仔细分析了每个模型生成的分子,并以测试集中的真实分子作为参考基准。当模型生成的分子亲和力优于参考分子时,我们将其结果标记为“优于参考”(BR)。分析表明,与其它模型相比,我们的模型表现出较低的多样性,这一发现正符合我们最初的预期。这种多样性下降的原因在于,在3DSMILES-GPT的训练阶段,我们施加了与物理化学性质相关的约束条件;同时,在分子生成过程中,又进一步限制了QED和log P值,最终导致生成分子的多样性显著降低。
我们还量化了3DSMILES-GPT生成的分子中,有多少分子的Vina评分低于参考分子。值得注意的是,3DSMILES-GPT生成的分子中有53%表现出比参考分子更低的Vina评分,而Pocket2Mol和TargetDiff则分别为48%。然而,尽管Lingo3DMol生成的分子通常具有比参考分子更高的亲和力,但这些分子在Vina评分上却集中分布在较窄的范围内(图3)。这种特性或许适用于某些药物发现任务,但如果参考分子本身普遍具有较高亲和力,例如针对ATK1和CDK2等激酶靶点时,BR分子的比例可能会相应下降。相比之下,3DSMILES-GPT展现出更强的能力,能够更广泛地探索化学空间,从而对目标表现出更高的结合亲和力——这正是我们模型的一大优势。此外,我们还计算了BR集合中分子的平均QED和SAS值(表1),结果显示,3DSMILES-GPT始终保持着优异的表现,尤其是在QED指标上,其提升幅度高达约33%。
总之,3DSMILES-GPT不仅具备生成具有更高结合亲和力分子的能力,还能显著改善QED和SAS指标。尤为突出的是,它能够直接生成与实际物理构象相当的分子结构,甚至无需经过重新对接步骤即可达到类似效果——这是其他模型所不具备的、直接基于物理构象生成的独特优势。
基于结构的药物设计,针对特定靶点
许多已报道的基于口袋的分子生成方法,缺乏在训练集之外的真实靶点上的测试,这使得人们对其在实际药物设计任务中的实用性产生质疑。为此,我们选取了四个与训练集和测试集均无关的蛋白质靶点:AKT1(4gv1)、SARS-COV2 3CL蛋白酶(7d3i)、CDK2(1h00)和DDR1(5bvk)。这些靶点已被广泛应用于虚拟筛选及分子生成任务,从而帮助我们模拟真实的药物发现场景。如图4a所示,3DSMILES-GPT生成的分子在上述四种不同蛋白家族的靶点口袋中,其Vina评分大多集中在−10至−5 kcal mol−1范围内。与Pocket2Mol和TargetDiff相比,这一分布更偏向横轴左侧,表明其具有更高的结合能和更强的亲和力。相比之下,Lingo3DMol生成的分子亲和力分布则主要集中在−10至−7之间,且更多聚焦于高亲和力区域,略逊于3DSMILES-GPT。不过,我们的模型在DDR1以外的其他靶点上,生成了更多高亲和力分子(Vina评分≤−10)。
然而,在实际药物发现过程中,高亲和力并非唯一追求的目标,同时还需要兼顾良好的成药性。因此,我们对所有分子进行了筛选,并重新分析了筛选后分子的Vina评分分布(见图4b)。结果显示,3DSMILES-GPT生成分子的Vina评分分布与筛选前基本一致,这表明绝大多数生成分子均符合我们的成药性标准。此外,其他基线模型生成的大部分高亲和力分子(Vina评分≤−10),在筛选过程中被成功过滤掉;而这一范围内的分子则主要由3DSMILES-GPT生成。在虚拟筛选阶段,由于可进行活性验证的分子数量有限,药物化学家通常会优先挑选少数几个对接评分较高的分子进行测试。由此可见,3DSMILES-GPT不仅能高效生成高亲和力分子,还能确保这些分子具备优良的成药性,这预示着该模型在实际药物发现应用中,有望助力开发出具有潜在活性、可在分子或细胞水平上通过验证的先导化合物。
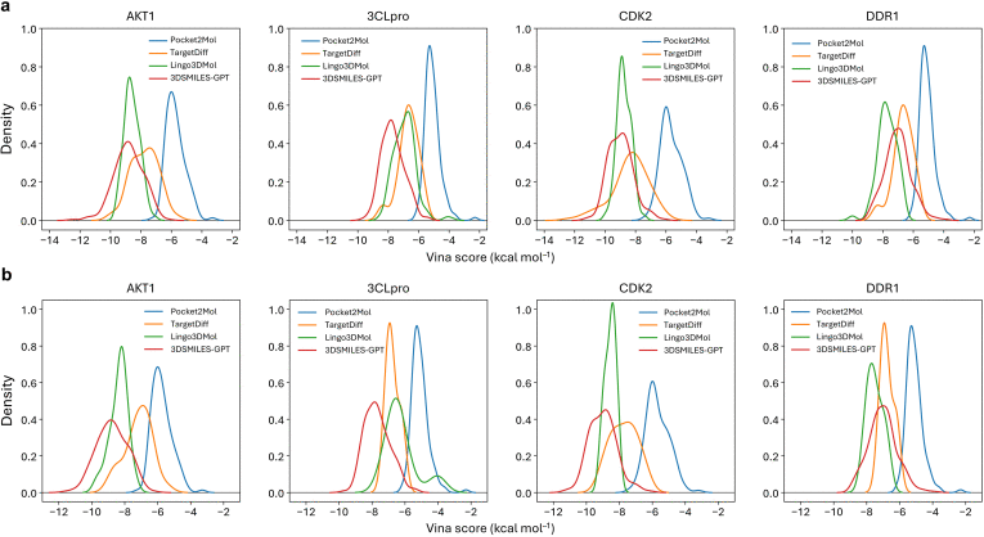
图4. 各特定靶点的Vina评分分布。(a)每个模型生成的所有分子的分布。(b)经过类药性筛选后(QED ≥ 0.3,SAS ≤ 5)的分布。左侧横轴表示亲和力越佳。
除了亲和力之外,在特定目标口袋中生成分子的目的,还涉及将所生成分子的结构与结合模式,与已知配体进行比较。如图5所示,我们选取了每种模型生成的亲和力最高的分子,以展示它们的结合模式。在基线模型中,Pocket2Mol和TargetDiff倾向于生成简单的芳香环衍生物或复杂的巨环化合物,但缺乏明确的目标特异性。而Lingo3DMol生成的分子则明显大于原始配体,这与测试集结果中获得的平均分子量480道尔顿相一致。此外,在CDK2口袋内的分子案例中,由于分子尺寸过大及键角不合理,出现了断裂现象,并与蛋白口袋发生碰撞(见图S1†)。相比之下,与其他基线模型相比,3DSMILES-GPT生成的分子结构更为合理,且其在口袋内的结合模式也更接近于原始配体。然而,在DDR1口袋中,这些分子的结合模式却偏向外部,这或许正是3DSMILES-GPT在DDR1靶点上未能显著优于其他基线模型的原因所在。
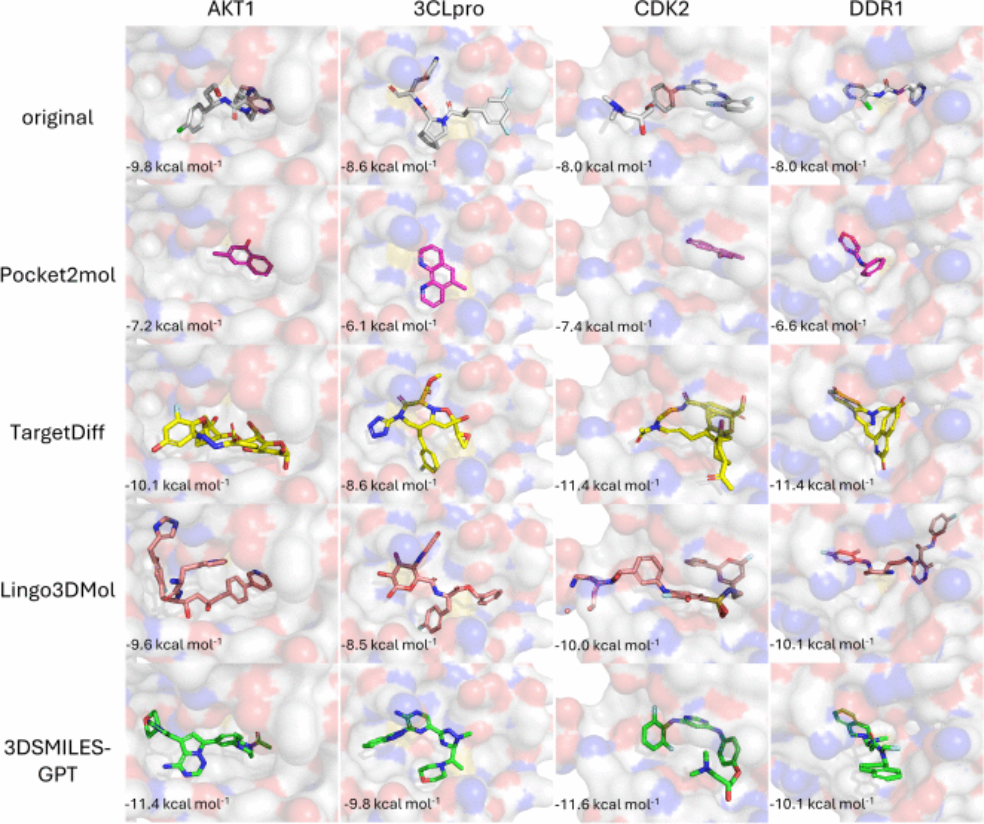
图5:各模型针对特定靶点生成的具有最佳亲和力的分子结合模式,以及与原始配体结合模式的比较。
总体而言,针对特定靶点口袋的测试表明,与先前的基线模型相比,3DSMILES-GPT能够生成更多符合药物相似性标准且对靶点具有高亲和力的分子,同时展现出更合理的结构,并对不同靶点表现出更高的结构特异性。
结论
在本研究中,我们提出了3DSMILES-GPT,这是一种创新的仅基于标记、具备口袋感知能力的分子生成方法,可针对蛋白质口袋内精准构建三维分子结构。该方法充分利用了大规模语言模型的强大功能,不仅能生成化学上有效的分子结构,还能确保这些分子展现出最佳的生物物理与化学特性。实验表明,3DSMILES-GPT在多项基准测试指标上表现卓越,尤其在生成具有更优Vina对接评分及更高药物相似性的分子方面,全面超越现有方法。尤为值得一提的是,所生成分子的QED值提升了33%,这表明我们的模型能够更高效地产出符合制药行业对候选药物标准要求的分子。此外,与当前最快的生成方法相比,3DSMILES-GPT的生成速度实现了三倍提升,完美满足了快速筛选药物候选物的实际需求。通过数据集外评估,进一步验证了该模型具备生成与特定靶点具有强结合力的类药分子的能力,充分展现了其在真实药物发现场景中的巨大潜力。未来,我们将以3DSMILES-GPT为基础,结合先进的大模型技术与海量训练数据,致力于开发一种通用的药物设计语言模型。总而言之,3DSMILES-GPT标志着药物发现领域分子生成方式的一次重大变革,它巧妙地利用大规模语言模型的强大能力,为应对复杂的生物学挑战提供了全新解决方案。
数据可用性
基于口袋的分子生成数据集已提供于:https://drive.google.com/drive/folders/1CzwxmTpjbrt83z_wBzcQncq84OVDPurM。
本研究中使用的代码已公开发布在GitHub仓库中:https://github.com/ashipiling/GPT_3DSMILES。