LIANA | part2 results部分
心血来潮想一并把results部分写完。
1 资源层:16 个配体-受体数据库到底“长啥样”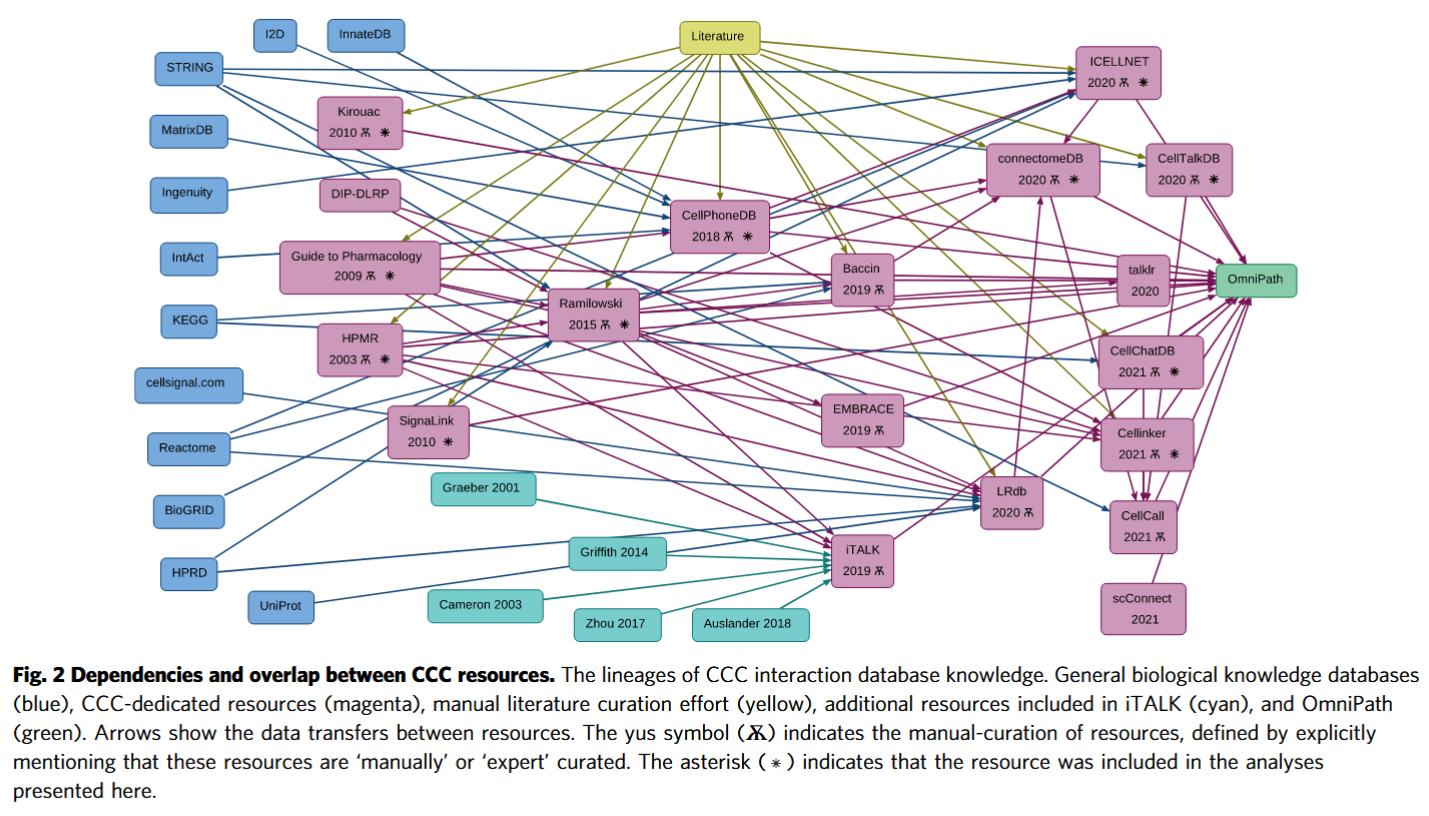
图 | 说人话 | 关键量化数字 |
|---|---|---|
Fig. 2 “族谱图” | 谁抄谁的一目了然:几乎所有资源都直接或间接抄了 Ramilowski2015(FANTOM5)那一版人工整理数据;再往上则是 KEGG、Reactome、STRING 这几个“爷爷级”通用库。[2,31,33–35] | 没给具体比例,只画依赖箭头。 |
Fig. 3A “有多少独一无二” | 平均每个库里:受体独有 6.4%、配体独有 5.7%、整张交互独有 10.4%。Cellinker 是个特例,39% 的交互别人家都没有。[Fig.3A;Suppl.Tab.1] | 独有交互最高 vs 均值:39% vs 10% |
Fig. 3B 热图 | 把 16 个库两两算 Jaccard(交集/并集)。ConnectomeDB、CellTalkDB、LRdb、iTALK 四个库相似度很高;而 CellPhoneDB、CellChatDB、Baccin、EMBRACE 自成一派。[Fig.3B;Suppl.Fig.S1] | 0.2–0.6 之间变化;>0.6 视为“高重合”。 |
补充观察 | 备注 | |
通路/功能偏向 Fig.4 & Suppl.Fig.S3–S11 | • RTK、JAK/STAT、WNT、Notch 信号通路基本各家都覆盖。 | |
组织富集蛋白 | 大部分库分布均匀,但乳腺、骨髓、淋巴结、下丘脑这几类组织明显被某些库忽视或偏爱。[Fig.4D] |
结论一句话:选不同的库,就等于从“不同视角”看细胞对话,功能解读会被带偏。
2 方法层:把 7 个算法 × 16 个库全部跑一遍看看到底差多少
图 | 说人话 | 关键数字 |
|---|---|---|
Fig. 5A 同一资源换方法 | 固定一个库,换打分方法,Top 1000 交互中位 overlap ≈ 0.08(Jaccard 指数)。 | 0.045–0.112 之间 |
Fig. 5B 同一方法换资源 | 固定方法,换库,中位 overlap ≈ 0.12。 | 0.085–0.132 之间 |
Suppl.Fig.S12 | 换成 Top1% 而不是 Top1000,结果差不多:依旧低的可怜。 | |
稳健性测试 Suppl.Fig.S13 | 结果一句话 | |
① 细胞亚采样 | 把细胞随机删 5%–40%,大部分方法排名纹丝不动 → 技术噪音不敏感。[Suppl.S13A] | |
② 标签混洗 | 故意把细胞类型标签打乱,排名掉得很厉害 → 前置聚类非常关键。[Suppl.S13B] | |
③ “精确污染” | 只往库里偷偷塞假交互,但保留真交互的高分区 → 高排名几乎不受影响。[Suppl.S13C] | |
④ “随机污染” | 全部交互随机替换 40% → 所有方法直接崩溃。[Suppl.S13D] |
3 用外部证据给预测打分:空间、蛋白质、细胞因子三路验证
① 空间信息 → 用 10× Visium, seqFISH, merFISH
图 | 关键发现 | 数字细节 |
|---|---|---|
Fig. 6B | 脑皮层数据里,所有方法(除 crosstalk scores)都“偏爱”空间相邻的互作;乳腺癌就没那么一致了。→ 组织结构越整齐,预测越可信。[14,49] | 脑皮层 OR(空间相邻 vs 随机)>2,乳腺癌 OR 只在 1.2–1.5。 |
Suppl.Fig.S15 seqFISH | NATMI、logFC Mean、Connectome 能重现 seqFISH 的高邻细胞对;merFISH 基因空间太少,基本无信号。 | — |
② 细胞因子活性签名 → 43 条 CytoSig 路线
图 | 关键发现 | 数字细节 |
|---|---|---|
Fig. 6A | 每个方法在 HER2+ / TNBC 两乳腺癌亚型中,其 Top 优先交互与“下游活性”显著正相关;当拉到全部预测时回归基线。说明高排名更可信。 | OR(配体-受体对应活性阳性/阴性) 随排名增加呈指数下降,到 5000 名次左右接近 1。 |
③ CITE-seq 蛋白丰度 → 受体是否真正“被表达”
做法 | 结果 |
|---|---|
z-score>1.645 的受体为“阳性”,用 AUROC 看方法能否优先把它选出来。 | 各方法 AUROC 均>0.7;CellChat/CellPhoneDB 在高特异区间略胜。[Suppl.Note3] |
4 共识排名:Robust Rank Aggregation
细节 | 说明 |
|---|---|
Fig. 1 示意图右侧绿色框 | 把七个方法的打分表拼在一起,用 RRA 算法做整合,自动生成“共识排名”。[17,65] |
在独立数据里 | 共识排名往往稳居前三,既不太激进也不太保守,可作为“稳妥版”结果输出。 |
结果一句话收束
Dimitrov(2022)[1] 用 6 套单细胞数据、128 种方法×资源组合跑通,告诉我们:
“数据库挑错了,算法再高级也白搭;算法不换,数据库换一半结果就面目全非。好在,它们总体上还能和空间邻接、蛋白丰度、细胞因子活性‘对得上’——所以多留个心眼、多用几种验证,比盲信单一结果是更安全的做法。”
基于这篇文章的系统比较,没有一个“一刀切”的最佳数据库和最佳方法,而是取决于你关注的目标。作者给出了以下结论和提示:
- 关于数据库
- OmniPath 在覆盖面上最广(见“Agreement with other modalities”部分),整合了 16 个资源且含有 10 余个额外来源,被作者默认为综合评分时使用的统一资源 [1]。
- 但数据库之间差异很大:
– Ramilowski、ConnectomeDB、CellTalkDB、LRdb 等包含 60 % 以上的共有相互作用,覆盖面重复度极高 [1, Fig. 3B]。
– CellPhoneDB、CellChatDB、EMBRACE 等更专注于包含蛋白复合体,如果你关心异源复合体介导的信号,优先这些资源 [1, Fig. 3B 与讨论]。
– Cellinker 有约 39 % 特有的相互作用,假如想挖掘新的潜在通讯对,可额外纳入[1, Fig. 3A]。
- 关于方法
- CellChat 和 CellPhoneDB 在“去噪/假阳性过滤”步骤上更严格,用置换检验给出 p-value 阈值,与细胞因子活性外检验吻合度最高 [1, Fig. 6A;图注中虚线指示过滤后的交互仍然显著]。
- Connectome、logFC Mean 和 Consensus 在与空间邻接信息的吻合度(尤其在结构清晰的鼠脑皮层数据)表现最稳健 [1, Fig. 6B]。
- SingleCellSignalR 在简单性和速度上有优势,但相对过滤不那么严格,容易出现负信号 [1, Fig. 6A, Supplementary Fig. S14]。
- Consensus(所有方法的 Robustrank aggregate 综合排名) 通常能减小单个方法的偏差,在跨数据集一致性上表现最佳 [1, Fig. 6B;讨论部分]。
- 实用建议
- 如果你更看重生物学信号验证和空间一致性:可用 OmniPath(最大资源)+ CellChat 或 CellPhoneDB(自带 p-value 过滤),再与 Consensus 并列汇报以增强可靠性。
- 如果你想全面捕捉潜在通讯:在 OmniPath 之外 补充 Cellinker,并用 Consensus 做最终交集或排名整合。
- 若数据集异质度高(如乳腺癌),建议同时跑 CellChat/CellPhoneDB 过滤版 + logFC Mean,观察空间或细胞因子活动的叠加结果,以减少假阴性/阳性 [1, 讨论部分]。
总结:Dimitrov 等 (2022) [1] 并没有指明唯一最佳组合,而是推荐“OmniPath + 多方法共识(特别是 CellChat/CellPhoneDB 与 Consensus 结合)”的策略,以兼顾覆盖面、统计严谨性和跨模态验证的一致性。
Dimitrov 等 (2022) [1] 之所以能够对 16 种先验知识资源与 7 种推断方法做“一对一”、“一对多”、“多对一”乃至“全组合”的系统比较,核心在于他们把资源和方法解耦,并用统一的框架 LIANA 做了一致性处理。以下几点概述了他们“具体怎么统一”的:
- 统一的数据接口
所有 16 个资源(例如 CellChatDB、CellPhoneDB、ConnectomeDB 等)经过格式转换后都变成相同的表格结构:一行记录一条“配体–受体”(或者更广义的发送蛋白–接收蛋白) 相互作用,列至少包含ligand、receptor(发送蛋白、接收蛋白)以及可选的复合体亚基信息[^Methods]。“All the CCC resources … were queried from OmniPath … apart from minor processing differences, such as removal of duplicates, updating to the latest gene symbols … removal of genes lacking reviewed UniProt IDs.”[^Methods]
- 统一的方法调用接口
LIANA 对 7 种方法(CellChat, CellPhoneDB, Connectome, logFC Mean, NATMI, SingleCellSignalR, Crosstalk score)做了函数级封装。外部用户只需一行代码即可调用任何资源-方法组合,而不必管各方法原先的输出格式或内部实现细节。
例如 CellPhoneDB 需要的“permuted p-value”步骤,CellChat 需要的“mediator”处理,都通过内建封装自动完成。 - 统一的下游后处理
为排除不同工具打分尺度差异,作者一律取排名前 1 000 的相互作用(或前 1 %,作为对照)再计算 Jaccard、富集、富度等指标,实现了“同一评判口径”。“To compare the overlap … we kept the 1,000 highest ranked interactions by default, including ties.”[^Overlap analysis]
- 统一的表达矩阵格式与阈值
无论原始数据是 Seurat 对象还是原生 CellChat 对象,都在输入前被转换成按 cluster 汇总后的伪bulk 表达矩阵;不同方法内部的“≥10 % 细胞表达”阈值、复合体亚基最低表达逻辑,也在封装时做了参数对齐,确保比较环境一致。“Whenever available, we used the recommended conversion method … Log-transformed counts were used when this was not done internally by the method.”[^Input specifics]
- 统一的基因 ID 与物种映射
人与鼠的基因符号统一到最新的 HGNC/MGI;所有小鼠数据在运行 OmniPath 资源前先通过 biomaRt 完成符号转换,避免物种名称造成的假阴性。“For murine datasets, we converted the OmniPath to murine symbols using the biomaRt package.”[^Agreement with other modalities]
通过以上五层“统一”,Dimitrov 等 (2022) [1] 得以在方法或资源互换时,只改变单一变量,从而明确“到底是资源差异还是方法差异主导了预测结果”。