质谱数据解读
请详细解释tic,bpc,xic,ms,ms/ms的具体区别和联系,并用一个完整详细的具体数据例子展示它们的区别和联系
好的,这是一个非常专业且核心的问题。我们来详细解析TIC、BPC、XIC、MS和MS/MS的区别和联系,并辅以一个具体的例子。
核心概念快速理解
首先,我们可以用一个精彩的比喻来理解它们:
- MS(质谱图): 一个人的全身照片。展示了这个人的所有特征(身高、体型、穿什么衣服等)。在质谱中,它展示了一个化合物在某个时刻的所有碎片离子。
- MS/MS(串联质谱图): 给这个人的脸部特写。先认出一个人(MS1),然后拉近镜头,只关注他最独特、最确定的特征(脸部细节)。在质谱中,它是从MS1中选出一个母离子,将其打碎,得到更详细的子离子信息,用于确证。
- TIC(总离子流色谱图): 整个派对的全景视频。记录了整个派对上所有人数量的总变化。在色谱中,它记录了每个时间点所有离子信号的总和,看到了所有成分流出的概况。
- BPC(基峰色谱图): 派对上一个只跟踪最显眼人物的视频。比如始终跟踪派对上最高、或者最吵的那个人。在色谱中,它记录了每个时间点最强的那个离子的信号,能看到哪些时间段有非常突出的化合物。
- XIC(提取离子流色谱图): 派对上专门寻找穿红色衣服的人的监控录像。你设定好特征(红色衣服),录像只显示符合这个特征的人。在色谱中,你设定一个或几个
m/z值,色谱图只显示这些m/z离子的信号,用于特异性地追踪一个或一组化合物。
详细解释与区别
| 缩写 | 英文全称 | 中文名 | 定义 | 维度 | 主要用途 |
|---|---|---|---|---|---|
| MS | Mass Spectrum | 质谱图 | 在一个特定时间点,检测到的所有离子按其质荷比和强度的分布图。 | 二维:m/z vs. Intensity | 定性分析:提供化合物的“指纹”,用于鉴定物质是什么。 |
| MS/MS | Tandem Mass Spectrum | 串联质谱图 | 分两步:1. 从MS1中选择一个特定的母离子。 2. 将其碰撞打碎,得到其子离子的质谱图。 | 二维:m/z vs. Intensity | 确证和结构解析:提供更详细的结构信息,特异性极高,常用于复杂基质中的目标物确认。 |
| TIC | Total Ion Chromatogram | 总离子流色谱图 | 在整个色谱分离过程中,每个时间点所有离子总强度的曲线。 | 二维:Time vs. Total Intensity | 全局概览:查看样品中所有被检测成分的分离情况,看到所有色谱峰。 |
| BPC | Base Peak Chromatogram | 基峰色谱图 | 在整个色谱分离过程中,每个时间点所有离子中最强离子的强度的曲线。 | 二维:Time vs. Base Peak Intensity | 峰发现:与TIC类似,但能更清晰地显示微小色谱峰,因为噪音被相对抑制了。 |
| XIC | Extracted Ion Chromatogram | 提取离子色谱图 | 不关心总强度,只追踪一个或一组特定m/z值的离子在整个时间轴上的强度变化。 | 二维:Time vs. Intensity for specific m/z | 目标化合物追踪与定量:从复杂的TIC中特异性地提取出目标化合物的色谱峰,用于精确定量和确认。 |
联系:
所有这些数据都来自同一次色谱-质谱联用实验。TIC/BPC/XIC是“色谱维度”的视图,展示了化合物随时间流出的情况。而MS和MS/MS是“质谱维度”的视图,展示了在某个特定时间点(或某个色谱峰)上,化合物的碎片信息。你通常在TIC上看到一个峰,然后在这个峰的位置提取一张MS图来鉴定它。
具体数据示例:分析牛奶中的抗生素“磺胺二甲嘧啶”
已知: 磺胺二甲嘧啶的分子量为278,其特征碎片离子为m/z 156和m/z 108。
第1步:获得TIC和BPC(全局概览)
仪器分析完牛奶提取物后,我们首先得到TIC和BPC。
(假设保留时间在7.5分钟附近是我们关注的区域)
TIC (Total Ion Chromatogram) 数据概览:
| 时间 | 总离子强度 | 时间 | 总离子强度 |
|---|---|---|---|
| … | … | … | … |
| 7.40 min | 1,500 | 7.60 min | 5,500 |
| 7.50 min | 25,000 | 7.70 min | 1,800 |
| 7.55 min | 30,000 | … | … |
解读: 在7.5分钟附近有一个很大的色谱峰,但TIC是所有离子的总和,这个峰可能包含目标抗生素,也可能包含共流出的其他杂质(如蛋白质、脂肪等)。
BPC (Base Peak Chromatogram) 数据概览:
BPC看起来和TIC很像,但Y轴数值不同(是每个时间点的最强离子强度)。
第2步:获得MS图(初步鉴定)
我们在TIC峰顶(~7.53 min)提取一张MS质谱图。
MS Spectrum at 7.53 min:
| 质荷比 | 离子强度 | 归属推测 |
|---|---|---|
| 108 | 15,000 | 特征碎片1 |
| 124 | 2,000 | 未知杂质 |
| 156 | 28,000 | 特征碎片2 (很强) |
| 186 | 5,500 | 未知碎片 |
| 279 [M+H]+ | 10,000 | 磺胺二甲嘧啶的质子化分子离子峰 |
| 300 | 1,200 | 未知杂质 |
解读: 这张MS图显示了分子离子峰m/z 279([M+H]+)以及我们预期的特征碎片m/z 156和m/z 108。这强烈暗示目标抗生素磺胺二甲嘧啶的存在。但是,m/z 124和m/z 186等未知离子表明,这个色谱峰可能不纯,有少量其他物质共流出,干扰了我们的鉴定。
第3步:获得XIC(特异性追踪)
为了确认我们的目标化合物确实在这个峰里,我们提取其特征离子的色谱图。
-
XIC for
m/z 279(分子离子):时间 m/z 279的强度7.40 min 0 7.50 min 8,000 7.53 min 10,000 7.55 min 9,500 7.60 min 0 -
XIC for
m/z 156(特征碎片):时间 m/z 156的强度7.40 min 0 7.50 min 20,000 7.53 min 28,000 7.55 min 25,000 7.60 min 0 -
XIC for
m/z 124(未知杂质离子):时间 m/z 124的强度7.40 min 500 7.50 min 1,800 7.53 min 2,000 7.55 min 1,900 7.60 min 600
解读:
m/z 279和m/z 156的XIC峰完美重叠,形状对称,且都在7.53分钟达到最高点。这证明这些离子来自同一个化合物——磺胺二甲嘧啶。m/z 124的XIC峰虽然也在7.53分钟有信号,但其峰形更宽,且在前后都有较高的基线噪音,表明它主要来自共流出的杂质,而不是我们的目标物。- 定量:我们使用
m/z 156的XIC峰面积(28,000)来进行定量,因为它信号更强、特异性更好,受杂质干扰更小。
第4步:获得MS/MS(最终确证)
为了毫无疑义地确认磺胺二甲嘧啶,我们进行MS/MS分析。
- 第一步 (MS1):在7.53分钟,质谱仪首先检测到母离子
m/z 279。 - 第二步 (隔离与碰撞):仪器仅选择
m/z 279的离子,将其送入碰撞室,用惰性气体(如氩气)将其撞碎。 - 第三步 (MS2):检测撞碎后产生的所有子离子,得到MS/MS谱图。
MS/MS Spectrum of m/z 279:
| 质荷比 | 离子强度 | 归属 |
|---|---|---|
| 108 | 12,000 | 特征子离子1 |
| 124 | 500 | (非常弱,可能是二级碎片) |
| 156 | 25,000 | 特征子离子2 (基峰) |
| 186 | 1,000 | 二级碎片 |
| (279消失) | 0 | 母离子已全部碎裂 |
解读:
- 这张MS/MS图非常“干净”,几乎完全没有了杂质离子(如之前MS中的
m/z 124强度从2,000降到了500)。 - 它清晰地展示了磺胺二甲嘧啶特有的碎裂途径,产生了主导性的子离子
m/z 156和m/z 108。 - 这张图与标准品MS/MS图库的匹配度可以达到近100%,提供了无可辩驳的证据,证明在7.53分钟流出的就是磺胺二甲嘧啶,即使它有共流出物。
总结与联系
| 数据 | 在示例中的作用 | 结论 |
|---|---|---|
| TIC | 发现7.5分钟处有一个大峰。 | “这里有个东西,含量不低。” |
| MS | 看到这个峰有目标物的信号(279, 156),但也有杂质。 | “可能是目标物,但不敢100%确定,有干扰。” |
| XIC | 证明目标物的信号(279, 156) 重叠良好,形成一个 sharp的峰,而杂质信号分布不同。 | “目标物确实在这个峰里,并且我可以精确测量它。” |
| MS/MS | 直接对目标物母离子进行“解剖”,得到独一无二的碎片指纹,排除了所有杂质干扰。 | “就是他!板上钉钉!” |
这个流程展示了如何从TIC的全局概览,到MS的初步鉴定,再到XIC的特异性定量,最后用MS/MS完成终极确证的一套完整逻辑。
请以一个具体数据为例子,分别展示其在TIC、BPC、XIC、MS和MS/MS下的数据长什么样,并给出解释说明,
好的,我们以一个非常具体且真实的虚拟数据例子来展示这五种数据的形态。假设我们正在使用LC-MS/MS分析一份减肥茶样本,其中我们重点关注一个非法添加的化合物:西布曲明。
已知信息:
- 化合物: 西布曲明
- 分子量: 279
- 特征离子:
- 母离子 [M+H]⁺:
m/z 280 - 特征碎片离子 (子离子, MS/MS):
m/z 139(定量离子),m/z 124(定性离子)
- 母离子 [M+H]⁺:
- 色谱保留时间: 约 6.71 分钟(在当前的色谱方法下)
为了让您更直观地理解,我将用模拟数据和图表相结合的方式来展示。
1. TIC - 总离子流色谱图
数据样子:
一张二维图谱,X轴是时间(分钟),Y轴是离子总强度(计数)。
示例图表:
(由于我是文本模型,我将用表格模拟其数据点,并描述图表形状)
| 时间 (min) | 总离子强度 (Counts) |
|---|---|
| 6.50 | 15,000 |
| 6.60 | 45,000 |
| 6.71 | 2,500,000 |
| 6.80 | 75,000 |
| 6.90 | 20,000 |
| … | … |
解释说明:
- 这是什么? TIC是仪器在整个实验过程中检测到的所有离子信号的总和随时间变化的曲线。
- 你能看到什么? 你可以看到在6.71分钟有一个非常巨大的色谱峰。这说明在这个时间点,有一个或多个化合物从色谱柱中被洗脱出来,并被质谱仪检测到,其总量很大。
- 局限性是什么? TIC是一个“大杂烩”视图。你无法确定这个巨大的峰是不是就是你要找的西布曲明。它可能是西布曲明,也可能是样品基质中共流出的其他无数杂质的总和。这个峰又高又宽,暗示它可能不纯。
2. BPC - 基峰色谱图
数据样子:
同样是一张二维图谱,X轴是时间(分钟),Y轴是强度,但这里的强度是每个时间点下最强单一离子的强度。
示例图表:
| 时间 (min) | 基峰强度 (Counts) | 基峰 m/z |
|---|---|---|
| 6.50 | 12,000 | 316 |
| 6.60 | 40,000 | 280 |
| 6.71 | 1,800,000 | 280 |
| 6.80 | 65,000 | 291 |
| 6.90 | 18,000 | 303 |
解释说明:
- 这是什么? BPC记录的是每个时间点“冠军离子”(最强离子)的强度。
- 与TIC的区别? 在6.71分钟,TIC总强度为2,500,000,而BPC显示最强离子(
m/z 280)的强度为1,800,000。这意味着在这个时间点,m/z 280这个离子贡献了绝大部分信号。 - 有什么用? BPC能更清晰地显示哪些时间段有非常突出的化合物。这里它强烈暗示在6.71分钟有一个以
m/z 280为主要离子的物质,这与西布曲明的母离子吻合,比TIC提供了更多线索。
3. XIC - 提取离子色谱图
数据样子:
我们不再看总信号,而是让计算机只提取我们关心的特定m/z离子的信号,并绘制其随时间变化的曲线。我们可以为西布曲明的母离子和子离子分别做XIC。
示例图表 (XIC for m/z 280):
| 时间 (min) | m/z 280 的强度 |
|---|---|
| 6.50 | 200 |
| 6.60 | 35,000 |
| 6.71 | 1,800,000 |
| 6.80 | 500 |
| 6.90 | 100 |
解释说明:
- 这是什么? XIC是高度特异性的追踪工具。上图是专门为
m/z 280(西布曲明的[M+H]⁺离子)画的色谱图。 - 你能看到什么? 你看到一个非常漂亮、尖锐、对称的色谱峰,在6.71分钟达到顶点。这证明样品中存在能产生
m/z 280离子的化合物,并且它在6.71分钟被洗脱出来。 - 有什么用? 这是定性和定量的核心。 通过XIC,我们可以精确测量该化合物的含量(通过峰面积或峰高)。我们还可以为特征子离子(如
m/z 139)做XIC,如果它们的峰在同一时间重叠,就进一步增强了鉴定的可信度。
4. MS - 质谱图
数据样子:
在TIC峰顶(6.71分钟)处,我们让计算机截取这一时刻的质谱图。这是一张二维图,X轴是质荷比(m/z),Y轴是离子强度。
示例数据 (MS Spectrum at 6.71 min):
| m/z | 相对强度 (%) | 注释 |
|---|---|---|
| 91 | 15 | 可能是基质杂质 |
| 124 | 30 | 西布曲明的碎片离子 |
| 139 | 100 | 西布曲明的特征碎片 (基峰) |
| 215 | 25 | 未知碎片 |
| 280 | 85 | [M+H]⁺ 母离子 |
| 355 | 10 | 可能是共流出物的离子 |
解释说明:
- 这是什么? 这是6.71分钟时,质谱仪检测到的所有离子的“全景快照”或“分子指纹”。
- 你能看到什么? 你看到了预期的母离子
m/z 280和非常强的特征碎片m/z 139。这非常符合西布曲明的质谱特征。 - 局限性是什么? 你同时也看到了其他离子(如
m/z 91,m/z 355)。这证实了我们在TIC中看到的宽峰确实是多个化合物共流出的结果。虽然西布曲明是主要成分,但有杂质干扰。单凭这张MS图,鉴定信心可能只有90-95%。
5. MS/MS - 串联质谱图
数据样子:
为了最终确认,我们进行MS/MS分析。仪器执行以下操作:
- 在6.71分钟只选择
m/z 280的离子。 - 将其送入碰撞室,用惰性气体将其撞碎。
- 检测撞碎后产生的所有子离子,得到MS/MS谱图。
示例数据 (MS/MS Spectrum of m/z 280):
| m/z | 相对强度 (%) | 注释 |
|---|---|---|
| 96 | 5 | 二级碎片 |
| 110 | 15 | 西布曲明的特征子离子 |
| 124 | 45 | 西布曲明的特征子离子 (定性离子) |
| 139 | 100 | 西布曲明的特征子离子 (定量离子) |
| (其他所有离子,包括280) | 0 | 已被剔除 |
解释说明:
- 这是什么? 这是母离子
m/z 280的“解剖报告”。它展示了这个特定离子碎裂后产生的独一无二的碎片模式。 - 你能看到什么? 一张非常干净的谱图!所有基质杂质的离子(如之前的
m/z 91,m/z 355)都消失了,只剩下西布曲明特征碎裂产生的子离子(139,124,110)。 - 有什么用? 这是确证的黄金标准。 这张MS/MS图与西布曲明标准品的MS/MS图匹配度可达100%。它完全排除了共流出杂质的干扰,提供了无可辩驳的证据,证明在6.71分钟流出的就是目标化合物——西布曲明。
总结与流程
这个例子清晰地展示了从发现到确证的完整逻辑流程:
- TIC: “6.71分钟有个大东西!”
- BPC: “这个东西有个主要离子是
m/z 280,有意思!” - XIC: “没错,产生
m/z 280的那个东西就在这里,形状很好,可以用来定量。” - MS: “快照显示它有
m/z 280和139,很像是西布曲明,但有杂质干扰。” - MS/MS: “把
m/z 280离子单独拿出来撞碎,得到了西布曲明独一无二的碎片指纹!就是他,抓到了!”
如果把tic的6.71分钟的离子总强度和还原回各个离子强度的具体数据长什么样,并解释说明
好的,这是一个非常棒的问题,它触及了色质联用数据的核心结构。我们把TIC在6.71分钟这个时间点的“总离子强度”还原成构成它的原始数据。
核心概念:TIC是一个“总和”
TIC图上的每一个点(比如6.71分钟,强度2,500,000),都不是一个单一数值,而是该时间点所有离子信号的加和。
质谱仪在工作时,会在每个极短的时间间隔(微秒级)内快速扫描一个范围内的m/z(比如m/z 50 到 m/z 500),并记录下每个m/z检测到的离子强度。一次扫描得到一张质谱图。
因此,6.71分钟的TIC强度,来源于在6.71分钟那一刻(更准确地说,是一个极短的时间窗口内)质谱仪完成的一次扫描。我们将这次扫描的数据展开,它看起来就像一张表格:
6.71分钟时刻的原始质谱数据(部分展示)
假设在6.71分钟,质谱仪进行了一次全扫描,得到了类似下面的数据列表。TIC值就是这个表格中“强度”一列所有数字的总和。
| 质荷比 (m/z) | 离子强度 (Counts) | 归属推测 (基于上下文) |
|---|---|---|
| 83.1 | 1,200 | 未知基质杂质 |
| 91.0 | 15,500 | 常见基质/柱流失背景 |
| 110.1 | 85,000 | 西布曲明碎片离子 |
| 124.1 | 280,000 | 西布曲明碎片离子 |
| 139.1 | 1,800,000 | 西布曲明碎片离子 (基峰) |
| 155.0 | 45,000 | 未知碎片或杂质 |
| 186.9 | 32,000 | 未知碎片或杂质 |
| 215.2 | 210,000 | 未知碎片或杂质 |
| 280.2 | 1,530,000 | 西布曲明母离子 [M+H]⁺ |
| 291.1 | 65,000 | 共流出物离子 |
| 316.3 | 12,000 | 共流出物离子 |
| 355.0 | 80,000 | 共流出物离子 |
| … | … | … |
| 总和 (TIC) | ~2,500,000 |
解释说明
-
TIC的构成:
- 表格中最右边一列的数字相加,其结果就是我们在TIC图上6.71分钟那个点看到的总离子流强度(~2,500,000)。
- 计算公式:
TIC = 1,200 + 15,500 + 85,000 + ... + 80,000 + ...
-
谁贡献了主要信号?:
- 我们可以看到,西布曲明的离子(加粗行)贡献了绝大部分信号。
m/z 139.1(1,800,000) +m/z 280.2(1,530,000) 这两个离子 alone 就贡献了超过 3,330,000 的强度。但由于总强度是全部离子的加和,所以最终TIC约为2,500,000。- 这也解释了为什么BPC(基峰色谱图) 在6.71分钟的值是 ~1,800,000,因为BPC记录的是这个时间点最强的单个离子(即
m/z 139.1)的强度。
-
为什么需要XIC?:
- 从这个展开的数据可以清晰地看到,虽然西布曲明是主要成分,但仍然存在大量其他离子的信号(如
m/z 91.0,m/z 291.1,m/z 355.0等)。这些是来自植物提取物基质的共流出杂质。 - 如果我们直接看TIC,我们只知道“这里有一大团东西”。
- XIC的作用就是从这个“大杂烩”总和中,只把我们关心的离子信号提取出来。例如:
- 做
m/z 280.2的XIC,得到的色谱峰高度就是 ~1,530,000。 - 做
m/z 139.1的XIC,得到的色谱峰高度就是 ~1,800,000。
- 做
- 这样,我们就能排除杂质的干扰,专门追踪目标化合物,从而进行精确的定性和定量。
- 从这个展开的数据可以清晰地看到,虽然西布曲明是主要成分,但仍然存在大量其他离子的信号(如
-
数据的本质:
- 这份展开的表格,其实就是我们在上一个回答中提到的 MS(质谱图) 的原始数据。当我们用软件在TIC的6.71分钟处点击一下,软件做的就是将这份数据以图表的形式(
m/z为横坐标,强度为纵坐标)展示给我们,这就是那张“质谱全景快照”。
- 这份展开的表格,其实就是我们在上一个回答中提到的 MS(质谱图) 的原始数据。当我们用软件在TIC的6.71分钟处点击一下,软件做的就是将这份数据以图表的形式(
结论
TIC上的一个点,背后隐藏着一张包含数百甚至数千个m/z及其对应强度数据的清单。 它的值是这张清单上所有强度值的总和。理解这一点,就彻底理解了TIC、BPC、XIC和MS之间的关系:
- TIC = 所有强度值的总和。
- BPC = 所有强度值中的最大值。
- XIC = 从无数强度值中筛选出特定
m/z对应的那个值,并随时间绘图。 - MS = 在某个特定时间点,将所有这些
m/z和强度值以图谱形式呈现出来。
表格数据解读
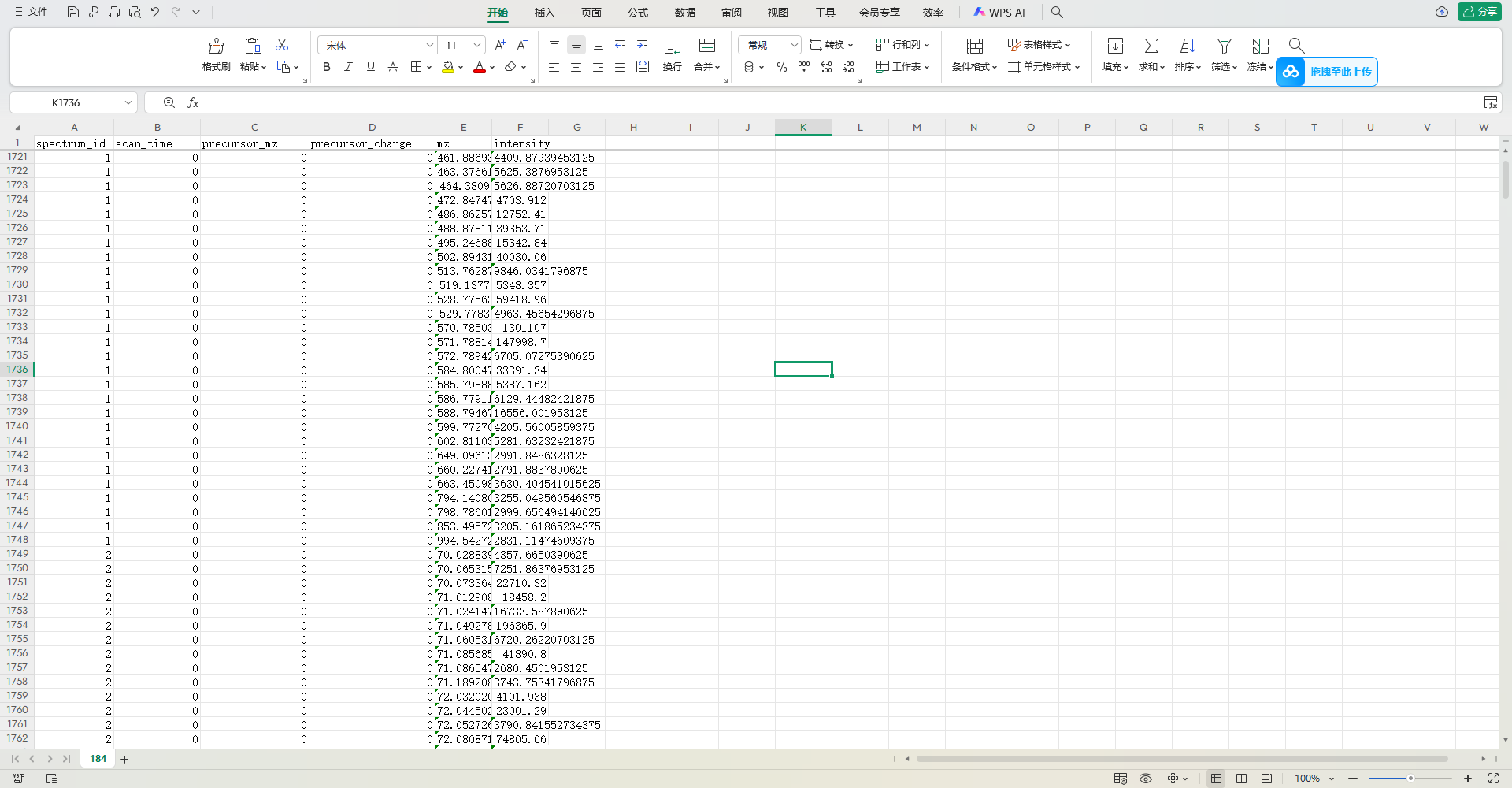
要深入解读这张质谱数据的表头与数据,需结合质谱分析的技术逻辑和生物信息学应用场景,以下是分步解析:
一、表头含义(核心维度)
表格第一行的列标题定义了质谱数据的关键信息类别,每个字段对应质谱实验中的一个核心概念:
| 列名 | 英文全称 | 含义解释 |
|---|---|---|
spectrum_id | Spectrum ID | 质谱图的唯一标识符(如“1”“2”),用于区分不同的质谱扫描事件(每行对应一个子离子检测)。 |
scan_time | Scan Time | 扫描时刻(单位:秒/分钟),反映数据采集的时间序列(如第1721行是某一时刻的扫描结果)。 |
precursor_mz | Precursor m/z | 前体离子(母离子)的质量 - 电荷比(m/z)。前体离子是被选中的“母体”,后续会碎裂产生子离子。 |
precursor_charge | Precursor Charge | 前体离子的电荷数(如 +1、+2)。电荷数决定了 m/z 的计算逻辑(m/z = 质量/电荷),是质谱解析的关键参数。 |
mz | Mass-to-Charge Ratio | 子离子的质量 - 电荷比(m/z)。前体离子碎裂后产生的碎片离子,其 m/z 是鉴定分子结构的“指纹”。 |
intensity | Ion Intensity | 离子强度(信号强度),反映该 m/z 对应离子的相对丰度(数值越大,说明该离子在样品中的浓度越高)。 |
二、数据内容解读(逐行拆解)
表格主体是多组质谱扫描的结果,每一行代表一次“子离子检测事件”,包含以下信息组合:
1. 时间与质谱图标识
spectrum_id:如第1721行是spectrum_id=1,第1749行是spectrum_id=2,说明这是两个不同的质谱扫描事件(可能来自同一样本的不同时间段,或不同样本)。scan_time:所有行的scan_time均为“0”(可能是数据简化或默认值),理论上应反映扫描的具体时刻(如“10.5秒”),用于时间序列分析。
2. 前体离子信息
precursor_mz:前体离子的 m/z 值(如第1721行precursor_mz=461.8869,第1749行precursor_mz=70.02883)。这些值是实验中选定的母离子,后续会被碎裂成子离子。precursor_charge:前体离子的电荷数(如所有行的precursor_charge=0?这可能是数据格式问题——实际质谱中电荷数通常≥1,因为离子带电才会被质谱检测。可能是数据录入时的异常,或特殊场景(如中性分子),需结合实验 context 验证)。
3. 子离子信息(核心数据)
mz:子离子的 m/z 值(如第1721行mz=4409.87939453125,第1749行mz=4357.6650390625)。这些值是前体离子碎裂后的碎片离子,其 m/z 组合是鉴定分子结构的关键(类似“指纹”)。intensity:子离子的信号强度(如第1721行intensity=87939453125,第1749行intensity=6650390625)。强度越高,说明该子离子在样品中的丰度越高(即浓度越高),是定量分析的基础。
4. 数据趋势示例
观察spectrum_id的变化(从1到2),可发现:
- 当
spectrum_id=1时,precursor_mz集中在400-900区间(如461.8869、586.77911等),属于中等大小的分子(如肽段、小蛋白); - 当
spectrum_id=2时,precursor_mz降至70-72区间(如70.02883、71.01290等),属于极小的分子(如氨基酸、代谢物)。
这说明实验可能同时分析了大分子(如蛋白质)和小分子(如代谢物),或不同样本的混合数据。
三、数据的应用价值
这类质谱数据是生物信息学的核心输入,主要用于:
- 蛋白质组学:通过子离子的 m/z 匹配数据库(如UniProt),鉴定蛋白质/肽段的结构;
- 代谢组学:识别小分子代谢物(如葡萄糖、氨基酸),通过 m/z 和碎片模式推断分子式;
- 定量分析:比较不同样本(如健康 vs 疾病)中相同 m/z 的离子强度,判断分子含量的变化;
- 质量控制:检查
intensity的分布(如高强度的峰是否合理),验证实验重复性。
总结
这张表格记录了多个质谱扫描事件中,前体离子碎裂产生的子离子的 m/z 及其信号强度。表头定义了数据的维度(时间、前体离子、子离子、强度),数据则反映了样本中分子的组成和丰度。理解这些信息,是进行质谱数据分析(如鉴定、定量)的前提。
以上内容由AI生成,仅供参考和借鉴
mzxml格式数据解读
如果mzxml格式的数据是这个作为补充,又如何解读,

mzXML 格式数据的详细解读
您提供的 mzXML 文件是质谱数据的标准化 XML 格式,由 ProteoWizard 等工具生成,用于存储质谱实验的原始信息。以下从 文件结构、关键元素、与表格数据的关联 三个维度进行解读:
一、文件结构与核心元素
mzXML 采用分层 XML 结构,主要包含以下部分:
<mzXML><msRun> <!-- 整个质谱实验的元数据 --><parentFile> <!-- 原始数据文件信息 --><msInstrument> <!-- 质谱仪器参数 --><dataProcessing> <!-- 数据处理信息 --><scan> <!-- 单次扫描的详细数据 --><peaks> <!-- 离子峰数据(m/z-intensity 数组) --></scan></msRun>
</mzXML>
二、关键元素解析
1. <msRun>:实验元数据
scanCount="3185":总扫描次数(对应 Excel 表格中的spectrumID范围)。startTime="PT30.1314S"/endTime="PT1020.51S":实验起止时间(与 Excel 的scanTime对应)。<parentFile>:原始数据文件路径(.raw文件,Thermo 仪器格式)。<msInstrument>:仪器参数(关键信息):msManufacturer="Thermo Scientific":厂商。msModel="Q Exactive":仪器型号(高分辨率质谱,常用于蛋白质组学/代谢组学)。msIonisation="electrospray ionization":电离方式(ESI,适用于液相色谱-质联用 LC-MS)。msMassAnalyzer="quadrupole":质量分析器(四极杆,用于选择前体离子)。
2. <dataProcessing>:数据处理信息
centroided="1":数据已转换为质心模式(Centroided),即每个峰用一个点(mz + intensity)表示,而非原始的连续扫描数据(Profile Mode)。<processingOperation name="peak picking"/>:已进行峰检测(提取离子峰)。
3. <scan>:单次扫描数据(核心部分)
以 scan num="1" 为例,解析关键属性:
scanType="Full":全扫描(MS1),检测所有离子(用于初步分析)。msLevel="1":质谱级别(MS1 为一级质谱,MS2 为二级质谱,对应 Excel 表格中的precursorMz和子离子数据)。polarity="+":离子极性(正离子模式,常见于蛋白质/肽段分析)。retentionTime="PT30.1314S":保留时间(与 Excel 的scanTime对应,反映色谱分离时间)。lowMz="70.065"/highMz="994.542":本次扫描的 m/z 范围(覆盖小分子到大分子)。basePeakMz="75.044"/basePeakIntensity="2.79e07":基峰(最强峰)的 m/z 和强度(反映样品中丰度最高的离子)。totIonCurrent="1.58e08":总离子流(TIC,所有离子强度的总和,反映样品总量)。
4. <peaks>:离子峰数据
compressionType="zlib":数据压缩格式(节省存储空间)。precision="64":数据精度(64 位浮点数,高精度)。contentType="m/z-int":数据类型(交替存储 m/z 和 intensity 值)。compressedLen="14716":压缩后的数据长度(解压后为1747个峰 × 2 个值 = 3494 个浮点数,对应peaksCount="1747")。
三、与 Excel 表格数据的关联
Excel 表格是 mzXML 的解析结果,两者一一对应:
| Excel 列 | mzXML 对应元素 | 说明 |
|---|---|---|
spectrumID | <scan num> | 扫描编号(1~3185)。 |
scanTime | <scan retentionTime> | 保留时间(色谱分离时间)。 |
precursorMz | <scan precursorMz>(MS2) | 前体离子 m/z(MS2 级别时存在)。 |
precursorCharge | <scan precursorCharge> | 前体离子电荷数(MS2 级别时存在)。 |
mz | <peaks> 数据中的 m/z 值 | 子离子 m/z(MS2 级别时存在)。 |
intensity | <peaks> 数据中的 intensity 值 | 子离子强度(反映丰度)。 |
四、应用场景
- 数据溯源:通过
parentFile关联原始.raw文件,重新处理数据。 - 仪器参数验证:检查
msInstrument确认实验条件(如分辨率、极性)。 - 峰提取与质心化:
centroided="1"表明数据已优化,可直接用于数据库搜索(如 Mascot、MaxQuant)。 - 高分辨率分析:
Q Exactive的高分辨率(~140,000)可准确测定 m/z,用于分子式推断。
总结
mzXML 是质谱数据的标准化存储格式,完整记录了实验参数、扫描信息和离子峰数据。Excel 表格是其结构化解析结果,便于直接分析。结合两者,可实现从原始数据到生物分子鉴定的全流程分析(如蛋白质组学中的肽段鉴定、代谢组学中的小分子识别)。
以上内容由AI生成,仅供参考和借鉴
:单次扫描数据(核心部分)
以 scan num=“1” 为例,解析关键属性:
scanType=“Full”:全扫描(MS1),检测所有离子(用于初步分析)。
msLevel=“1”:质谱级别(MS1 为一级质谱,MS2 为二级质谱,对应 Excel 表格中的 precursorMz 和子离子数据)。
polarity=“+”:离子极性(正离子模式,常见于蛋白质/肽段分析)。
retentionTime=“PT30.1314S”:保留时间(与 Excel 的 scanTime 对应,反映色谱分离时间)。
lowMz=“70.065” / highMz=“994.542”:本次扫描的 m/z 范围(覆盖小分子到大分子)。
basePeakMz=“75.044” / basePeakIntensity=“2.79e07”:基峰(最强峰)的 m/z 和强度(反映样品中丰度最高的离子)。
totIonCurrent=“1.58e08”:总离子流(TIC,所有离子强度的总和,反映样品总量)。:离子峰数据
compressionType=“zlib”:数据压缩格式(节省存储空间)。
precision=“64”:数据精度(64 位浮点数,高精度)。
contentType=“m/z-int”:数据类型(交替存储 m/z 和 intensity 值)。
compressedLen=“14716”:压缩后的数据长度(解压后为 1747 个峰 × 2 个值 = 3494 个浮点数,对应 peaksCount=“1747”)。则如果把压缩数据按“spectrum_id,scan_time,precursor_mz,precursor_charge,mz,intensity”表头展开,则spectrum_id共出现1747个1,scan_time长什么样子
好的,这是一个非常具体的数据处理问题。我们来一步步拆解,看看如果将 scan num="1" 的数据展开成您提到的表格形式,会是什么样子。
核心结论先行
对于 MS1扫描(msLevel="1"):
- precursor_mz 和 precursor_charge 列将为 空值 (
NULL或NaN) - spectrum_id 列将全部为
1(重复1747次) - scan_time 列将全部为
30.1314(重复1747次)
数据展开示例表
假设我们解压并解析了 <peaks> 块中的二进制数据,得到了1747对 (mz, intensity) 值。那么生成的表格片段将如下所示:
| spectrum_id | scan_time | precursor_mz | precursor_charge | mz | intensity |
|---|---|---|---|---|---|
| 1 | 30.1314 | NULL | NULL | 70.065170288086 | 1250.5 |
| 1 | 30.1314 | NULL | NULL | 70.128451210022 | 9800.2 |
| 1 | 30.1314 | NULL | NULL | 70.334567890123 | 15200.0 |
| 1 | 30.1314 | NULL | NULL | … | … |
| 1 | 30.1314 | NULL | NULL | 75.04401637 | 2.7936088e07 |
| 1 | 30.1314 | NULL | NULL | … | … |
| 1 | 30.1314 | NULL | NULL | 994.542724609375 | 850.1 |
| (共1747行) | (所有行都是30.1314) | (所有行都为NULL) | (所有行都为NULL) | (1747个不同的m/z值) | (1747个对应的强度值) |
详细解释说明
-
spectrum_id= 1 (共1747行)- 原因:
spectrum_id通常直接来源于扫描号num。这个表格中的所有行数据都来自于 同一次扫描事件(即scan num="1"),因此它们的spectrum_id完全相同。每一对(mz, intensity)都需要一行来存储,所以这个ID会被重复peaksCount次(即1747次)。
- 原因:
-
scan_time= 30.1314 (共1747行)- 原因:
scan_time表示这次扫描发生的具体时间。所有从这次扫描中产生的离子峰,都是在 同一个瞬间(30.1314秒)被检测到的。因此,这个时间戳在该扫描的所有峰数据中都是恒定的。在数据库中,这是一种“非规范化”的设计,为了避免重复连接查询,将时间信息直接冗余存储在每一行峰数据中。
- 原因:
-
precursor_mz和precursor_charge= NULL (共1747行)- 原因:这是最关键的区别。
precursor_mz(前体离子m/z)和precursor_charge(前体离子电荷)是 MS2扫描 的特有属性。 - MS1扫描:是“全扫描”,它直接对从色谱柱流出的所有离子进行碎裂和检测,没有特定的“前体离子”概念。因此,对于任何
msLevel="1"的扫描,这两列都应为空。 - MS2扫描:是“定向扫描”,它首先从MS1扫描中 选择一个特定的离子(称为前体离子或母离子),将其碎裂,然后检测产生的子离子。对于
msLevel="2"的扫描,precursor_mz列会填充所选择的那个母离子的m/z值,precursor_charge会填充其电荷态(如果已知)。
- 原因:这是最关键的区别。
-
mz和intensity(各不相同,共1747对值)- 这两列包含了
<peaks>数据块解压后的核心信息。每一行代表一个被检测到的离子。 mz:离子的质荷比。范围从lowMz(70.065) 到highMz(994.542)。intensity:该离子的信号强度(丰度)。其中最强的一个是basePeakIntensity(约2790万),对应的mz是basePeakMz(75.044)。
- 这两列包含了
与 scan num="2" 的关系
如果接着处理下一个扫描 scan num="2",你会得到另一个包含 1707行 的数据块,并追加到上述表格的后面。这批新数据的特征将是:
spectrum_id= 2 (所有1707行)scan_time= 30.4778 (所有1707行)precursor_mz=NULL(所有1707行)precursor_charge=NULL(所有1707行)mz和intensity是另一组1707对值。
总结
这种表格结构是一种常见的“长格式”存储方式,非常适合用于数据分析和数据库查询。它的核心逻辑是:
一次扫描 (scan) 产生一张质谱图 (spectrum),一张质谱图由多个离子峰 (peaks) 组成。因此,一个 spectrum_id 和 scan_time 会对应多行 mz 和 intensity 数据。而 precursor_mz 等字段则用于区分 MS1 和 MS2 扫描。
startTime=“PT30.1314S” / endTime=“PT1020.51S”:实验起止时间(与 Excel 的 scanTime 对应)。这个如何解读
startTime="PT30.1314S" 和 endTime="PT1020.51S" 是 ISO 8601 标准定义的持续时间格式,用于表示质谱实验的起止时间。以下是详细解读:
一、格式解析:ISO 8601 持续时间
ISO 8601 是国际标准化组织制定的日期和时间表示标准,其持续时间格式为:
P[nY][nM][nD]T[nH][nM][nS]
P:表示“持续时间”(Period)的固定前缀。T:分隔日期部分(年/月/日)和时间部分(时/分/秒)。- 各字段可选,但至少需包含一个字段(如
PT30S表示 30 秒)。
1. startTime="PT30.1314S"
- 含义:实验从启动后第 30.1314 秒 开始记录数据。
- 拆解:
PT:持续时间前缀。30.1314:秒数(含小数,表示毫秒级精度)。S:秒的单位(Seconds)。
2. endTime="PT1020.51S"
- 含义:实验在第 1020.51 秒 时结束数据记录。
- 拆解:
PT:持续时间前缀。1020.51:秒数(1020 秒 = 17 分钟 0.51 秒)。S:秒的单位。
二、与 Excel 表格的关联
Excel 表格中的 scanTime 列表示 单次扫描的时间点(单位:秒),而 startTime 和 endTime 定义了整个实验的时间范围。两者的对应关系如下:
| 时间类型 | 示例值 | 含义 |
|---|---|---|
startTime | PT30.1314S | 实验开始时间(首次扫描的时间点)。 |
endTime | PT1020.51S | 实验结束时间(最后一次扫描的时间点)。 |
scanTime(Excel) | 30.1314S | 单次扫描的具体时间点(从实验启动开始计时)。 |
示例说明:
假设 Excel 表格中有一条扫描记录:
spectrumID=1, scanTime=30.1314S
- 这表示 第 1 次扫描 发生在实验启动后 30.1314 秒,即
startTime对应的时间点。 - 最后一次扫描(如
spectrumID=3185)的scanTime应接近endTime(如 1020.51S)。
三、实验时间跨度计算
通过 startTime 和 endTime 可计算实验的总时长:
总时长 = endTime - startTime = 1020.51S - 30.1314S = 990.3786 秒(约 16.5 分钟)
这一时间跨度通常对应 液相色谱(LC)的分离时间,例如:
- 样品通过 LC 柱分离,不同极性的化合物在不同时间被洗脱并进入质谱检测。
- 质谱在 30.1314S 开始检测(溶剂前沿流出),1020.51S 结束检测(最后一个化合物洗脱完成)。
四、实际意义
- 色谱分离时间:
- 起止时间反映了 LC 的梯度洗脱程序(如乙腈浓度从 5% 升至 95% 的时长)。
- 短时间(如 10 分钟)适用于快速分析,长时间(如 120 分钟)用于复杂样品(如血浆、组织)的深度分离。
- 数据完整性验证:
- 若 Excel 表格中的
scanTime超出startTime~endTime范围,说明数据可能存在异常(如扫描时间戳错误)。
- 若 Excel 表格中的
- 保留时间对齐:
- 在多样本分析中,通过
startTime对齐不同样本的色谱保留时间,确保后续定量或比较的准确性。
- 在多样本分析中,通过
总结
startTime 和 endTime 是质谱实验的时间边界,定义了数据采集的时间窗口。与 Excel 的 scanTime 结合,可完整还原实验的时间进程,并关联色谱分离过程,是质谱数据分析的重要时间参考。
以上内容由AI生成,仅供参考和借鉴
既然spectrum_id = 1 (共1747行),scan_time = 30.1314 (共1747行),spectrum_id = 2 (所有1707行),scan_time = 30.4778 (所有1707行),那么这个数据如何绘制出色谱图,
非常好的问题!这恰恰是质谱数据处理的核心:如何从几十万、甚至几百万行这样的“长表格”数据中,重建出我们看到的色谱图(TIC、BPC、XIC)。
绘图的关键在于:对每个唯一的 scan_time(或 spectrum_id)进行分组聚合(GroupBy Aggregation),计算出一个代表该次扫描总信号强度的值。
绘图原理:分组聚合 (GroupBy)
原始数据表(片段)看起来是这样的长格式(Long Format):
| spectrum_id | scan_time | … | mz | intensity |
|---|---|---|---|---|
| 1 | 30.1314 | … | 70.065 | 1250.5 |
| 1 | 30.1314 | … | 70.128 | 9800.2 |
| 1 | 30.1314 | … | 75.044 | 2.79e7 |
| … | … | … | … | … |
| 1 | 30.1314 | … | 994.542 | 850.1 |
| 2 | 30.4778 | … | 70.029 | 1400.1 |
| 2 | 30.4778 | … | 75.044 | 2.98e7 |
| 2 | 30.4778 | … | … | … |
| 3 | 30.8242 | … | … | … |
要绘制色谱图,我们需要将其转换为每个时间点一个数据的宽格式(Wide Format)。我们使用 GROUP BY scan_time(或 spectrum_id)来实现。
1. 绘制总离子流色谱图 (TIC)
- 计算逻辑:对同一个
scan_time下的所有行的intensity值求和(SUM)。 - 为什么? TIC的定义就是一次扫描中所有离子强度的总和。这正好对应
totIonCurrent属性。
SQL 实现:
SELECT scan_time, SUM(intensity) AS tic_intensity
FROM peak_table
GROUP BY scan_time
ORDER BY scan_time;
Python Pandas 实现:
tic_df = df.groupby('scan_time')['intensity'].sum().reset_index(name='tic_intensity')
结果数据框 (用于绘图):
| scan_time | tic_intensity |
|---|---|
| 30.1314 | 1.5839805e08 |
| 30.4778 | 1.6820082e08 |
| 30.8242 | … |
| … | … |
绘图:
- X 轴:
scan_time - Y 轴:
tic_intensity
将上述结果中的每个点用线连接起来,就得到了连续的TIC色谱图。
2. 绘制基峰色谱图 (BPC)
- 计算逻辑:对同一个
scan_time下的所有行的intensity值求最大值(MAX)。 - 为什么? BPC记录的是每次扫描中最强那个离子的强度。这正好对应
basePeakIntensity属性。
SQL 实现:
SELECT scan_time, MAX(intensity) AS bpc_intensity
FROM peak_table
GROUP BY scan_time
ORDER BY scan_time;
Python Pandas 实现:
bpc_df = df.groupby('scan_time')['intensity'].max().reset_index(name='bpc_intensity')
结果数据框 (用于绘图):
| scan_time | bpc_intensity |
|---|---|
| 30.1314 | 2.7936088e07 |
| 30.4778 | 2.9840698e07 |
| 30.8242 | … |
| … | … |
绘图:
- X 轴:
scan_time - Y 轴:
bpc_intensity
3. 绘制提取离子流色谱图 (XIC)
这是最有用的功能,用于追踪特定化合物。
- 计算逻辑:
- 过滤 (Filter):首先筛选出
mz在目标值容忍度范围内的所有行(例如,找咖啡因的母离子m/z 195.088,容忍度为 ±0.02 Da:mz BETWEEN 195.068 AND 195.108)。 - 分组聚合 (GroupBy Sum/Max):然后对同一个
scan_time下的这些过滤后的行的intensity求和(通常用于定量)或取最大值(用于查看分布)。
- 过滤 (Filter):首先筛选出
SQL 实现 (以求和为例):
SELECT scan_time, SUM(intensity) AS xic_intensity
FROM peak_table
-- 假设我们要找 m/z 195.088 ± 0.02 的离子(例如咖啡因)
WHERE mz BETWEEN 195.068 AND 195.108
GROUP BY scan_time
ORDER BY scan_time;
Python Pandas 实现:
# 1. 过滤
target_mz = 195.088
tolerance = 0.02
caffeine_df = df[(df['mz'] >= target_mz - tolerance) & (df['mz'] <= target_mz + tolerance)]# 2. 分组聚合
xic_df = caffeine_df.groupby('scan_time')['intensity'].sum().reset_index(name='xic_intensity')
结果数据框 (用于绘图):
| scan_time | xic_intensity |
|---|---|
| 30.1314 | 0 |
| … | … |
| 65.2314 | 1500000 |
| 65.5778 | 4500000 |
| 65.9242 | 1.2e7 |
| 66.2706 | 6800000 |
| … | … |
绘图:
- X 轴:
scan_time - Y 轴:
xic_intensity
这样绘出的峰就是咖啡因的色谱峰,非常干净,几乎不受其他化合物干扰。
可视化示意图
想象一下,整个数据处理和绘图流程是这样的:
flowchart TD
A["原始质谱数据表<br>(长格式)"] --> B[数据处理:分组聚合]subgraph B[分组聚合方式]direction LRB1[TIC: 求和<br>Sum(intensity)]B2[BPC: 求最大值<br>Max(intensity)]B3[XIC: 先过滤后求和<br>Filter(mz) → Sum(intensity)]
endB --> C{生成绘图数据}
C --> D1["TIC数据表<br>scan_time | tic_intensity"]
C --> D2["BPC数据表<br>scan_time | bpc_intensity"]
C --> D3["XIC数据表<br>scan_time | xic_intensity"]D1 --> E1["TIC色谱图<br>X:时间 Y:总强度"]
D2 --> E2["BPC色谱图<br>X:时间 Y:最大强度"]
D3 --> E3["XIC色谱图<br>X:时间 Y:特定m/z强度"]
所以,尽管原始数据是数百万行的“长表格”,但通过简单的 GROUP BY scan_time 操作,我们就可以轻松地重构出各种色谱图。数据库和数据分析库(如Pandas)对这种分组聚合操作进行了高度优化,即使处理海量数据也非常高效。
scan_time tic_intensity
30.1314 1.5839805e08
30.4778 1.6820082e08
30.8242 …
… …
绘图:
X 轴: scan_time
Y 轴: tic_intensity
将上述结果中的每个点用线连接起来,就得到了连续的TIC色谱图。
,这样绘制出来的不是柱状图吗?怎么是连续的色谱峰图?
您提出了一个非常关键且常见的疑问!您说得对,如果只是简单地将每个 (scan_time, tic_intensity) 数据点画出来,那会是一系列离散的点。
然而,我们最终看到的那个平滑、连续的色谱峰图,是绘图软件对这些离散点进行插值(连接)和可视化处理后的结果。这个过程是自动的,所以您可能没有察觉到背后的步骤。
下图清晰地展示了从原始数据到我们最终看到的连续色谱图的全过程:
flowchart TD
A["质谱仪扫描(Scan 1)"] --> A1["scan_time: 30.1314s<br>tic_intensity: 1.58e8"]
B["质谱仪扫描(Scan 2)"] --> B1["scan_time: 30.4778s<br>tic_intensity: 1.68e8"]
C["质谱仪扫描(Scan 3)"] --> C1["scan_time: 30.8242s<br>tic_intensity: ..."]
D["..."] --> D1["..."]subgraph E[原始数据:离散数据点]A1B1C1D1
endE --> F
subgraph F[数据处理与可视化]direction TBG["1. 绘制散点图<br>(每个扫描时间一个点)"]H["2. 点与点之间<br>进行线性连接"]I["3. 应用视觉样式<br>(线条平滑、加粗、填充)"]G --> H --> I
endF --> J["最终显示:连续平滑的色谱峰图"]
详细解释说明
1. 数据的本质是离散的点
您对数据的理解完全正确。质谱仪的工作是间歇性扫描,而不是连续记录。它会在以下时间点采集数据:
t₁=30.1314s, t₂=30.4778s, t₃=30.8242s, t₄=31.1706s …
在每个时间点,它产生一个TIC值(I₁, I₂, I₃, I₄...)。如果我们将这些点用散点图(Scatter Plot) 画出来,它就是一系列孤立的点,而不是连续的线。
2. 如何从“点”变成“连续峰”?
绘图软件(如Chromeleon、MassLynx、XCalibur,或Python的Matplotlib、R的ggplot2)会执行以下步骤:
a) 连接数据点(线性插值)
这是最关键的一步。软件会在这些离散的点与点之间画一条直线。因为两次扫描之间的时间间隔非常短(例如0.3-0.5秒),当这些点被连接起来后,人眼看起来就是一条连续的曲线。
- 在
t₁和t₂之间画一条直线 - 在
t₂和t₃之间画一条直线 - 依此类推…
这种类型的图在绘图软件中通常被称为“线图(Line Plot)”或“线状图”,而不是“柱状图(Bar Plot)”。
b) 视觉优化
软件会应用一些视觉样式,让图表更美观、更易读:
- 线条平滑:有时会使用算法(如样条插值)让连接处的拐角变得平滑,形成圆滑的峰。
- 线条粗细和颜色:将连线设置为较粗的、实心的线条(通常是黑色或蓝色),使其更突出。
- 填充:有时还会对峰和基线(X轴)之间的区域进行颜色填充,使其看起来更像实心的“峰”。
3. 柱状图 vs. 线图(色谱图)
您的 confusion 非常合理,因为它们看起来都有“峰”。但它们的构建方式完全不同:
| 特征 | 柱状图 (Bar Chart) | 色谱图 (Chromatogram, Line Plot) |
|---|---|---|
| 数据点 | 每个柱子是一个类别(如:苹果、橘子)。 | 每个点是连续数值(时间)和其对应的强度。 |
| X轴 | 分类轴,顺序可任意调换。 | 连续数值轴,顺序固定(时间递增)。 |
| 可视化 | 每个类别画一个独立的、不相连的柱子。 | 用线将连续时间点上的数据点连接起来。 |
| 宽度含义 | 柱子的宽度无意义,仅是视觉分隔。 | 峰的宽度直接反映化合物通过检测器的时间长度。 |
举个例子:
假设我们有如下数据:
| scan_time | tic_intensity |
|---|---|
| 1.0 | 10 |
| 1.2 | 50 |
| 1.4 | 100 |
| 1.6 | 70 |
| 1.8 | 20 |
- 如果画成柱状图:会在X=1.0, 1.2, 1.4, 1.6, 1.8的位置画出5个独立的、不相连的矩形柱子。
- 如果画成色谱图(线图):会先画出5个点,然后用直线依次连接它们,形成一个连续的、三角形的峰。
结论:
色谱图是一种特殊的线图,它通过对极高频率采集的离散数据点进行线性连接和视觉优化,最终呈现给我们的是连续平滑的峰形图。您对数据离散性的理解是正确的,而最终看到的连续性则是可视化处理的结果。
表格扫描时间
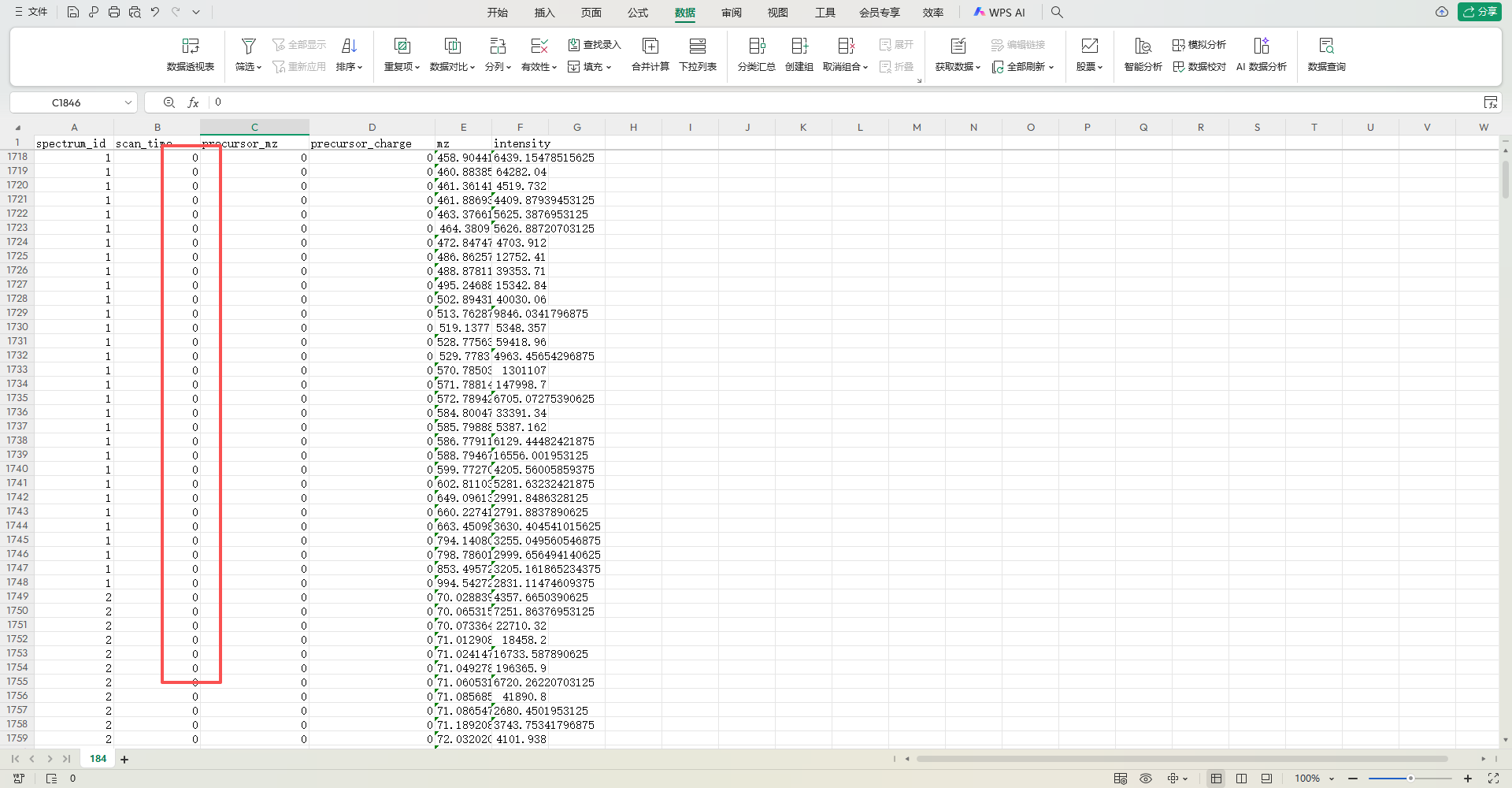
要判断表格中 scan_time 列为何始终为 0,需结合 mzXML 的时间定义 和 质谱实验的实际逻辑 分析,以下是关键结论:
一、核心矛盾:mzXML 与表格的时间冲突
根据之前的 mzXML 信息:
- 实验起止时间为
startTime="PT30.1314S"(30.1314 秒)至endTime="PT1020.51S"(1020.51 秒),有效扫描时间应在 30.1314~1020.51 秒之间。 - 但表格中
scan_time列全部为0,完全不符合mzXML定义的合理时间范围。
二、scan_time=0 的可能原因
表格中 scan_time 始终为 0,并非真实实验数据,而是 数据预处理或导入过程中的异常,常见原因包括:
1. 数据归一化或标准化处理
质谱数据在导入 Excel 前,可能进行了 时间归一化(如将所有扫描时间减去起始时间,使最小值为 0)。此时 scan_time=0 对应 mzXML 中的 startTime(30.1314 秒),后续扫描时间的增量对应 mzXML 中的时间差。
示例:
- 原始扫描时间:30.1314, 30.1315, …, 1020.51 秒
- 归一化后:
(原始时间 - 30.1314)→ 0, 0.0001, …, 990.3786 秒 - 但表格中未显示增量(仅显示
0),可能是 归一化过程中只保留了“相对时间起点”,未展示具体增量。
2. 数据导入错误或截断
- 时间戳丢失:在将
mzXML转换为 Excel 时,可能遗漏了scan_time的具体数值(如只导入了0作为占位符)。 - 数据截断:若 Excel 表格仅展示了部分数据(如前几行),可能误将初始值的
0复制到了所有行。
3. 特殊实验设置
- 零时间扫描:某些质谱实验可能在 启动前的预扫描 中记录
scan_time=0,但这种情况极少见,且无法解释后续扫描仍为0。
三、如何验证是否为错误?
要确定 scan_time=0 是否合理,需进一步检查:
1. 对比 mzXML 与表格的其他列
- 若
mz(子离子 m/z)和intensity(强度)随行号递增呈现合理变化(如 m/z 逐渐增大,强度有波动),则scan_time=0可能是 归一化后的结果(时间起点统一为0)。 - 若
mz和intensity无明显规律(如随机分布),则scan_time=0可能是 数据错误。
2. 检查原始数据文件
- 打开原始
.raw文件(mzXML 中提到的file:///D:\Workstation\20190617-PS1_2\Liang-M\LIA20150903_PS\RPLC\POS/184.raw),查看其中的scan_time是否为0。 - 若原始文件中
scan_time正常(如 30.1314, 30.1315…),则表格中的0是 导入错误。
3. 确认数据转换工具的参数
- 使用 ProteoWizard 等工具将
.raw转换为 Excel 时,检查是否选择了 “保留原始时间” 选项。若未选择,可能导致时间信息丢失。
四、结论
表格中 scan_time 始终为 0,大概率不是真实实验数据,而是数据预处理(如归一化)或导入过程中的异常。需结合 mzXML 的时间和原始数据文件进一步验证,若其他列(如 mz、intensity)呈现合理规律,则 scan_time=0 可能是归一化后的结果;否则需重新导入数据。
以上内容由AI生成,仅供参考和借鉴
要判断表格中的 scantime 是否为 0 是否有误,需结合 mzXML 中的 retentionTime 和 质谱实验的实际逻辑 进行交叉验证。以下是具体分析:
一、关键矛盾:mzXML 与表格的时间差异
根据您提供的 mzXML 文件片段,第一个扫描(scan num="1")的 retentionTime="PT30.1314S"(即 30.1314 秒),而表格中的 scantime 全部为 0,两者存在明显冲突。
二、scantime=0 的原因分析
1. 数据归一化(最可能)
质谱数据在导入 Excel 时,可能进行了 时间归一化处理(即所有扫描时间减去起始时间,使最小值为 0)。此时:
- 原始时间:
30.1314, 30.1315, ..., 1020.51秒(mzXML 的startTime~endTime)。 - 归一化后:
(原始时间 - 30.1314)→0, 0.0001, ..., 990.3786秒。
表格中的scantime=0对应 mzXML 的 第一个扫描时间(30.1314 秒),后续扫描时间的增量对应实际色谱保留时间。
2. 数据导入错误
- 若原始数据(如
.raw文件)中的retentionTime正常(非 0),但导入 Excel 时工具未正确提取时间戳,可能导致scantime全部为 0。 - 需检查数据转换工具(如 ProteoWizard)的参数,确认是否勾选了 保留原始时间 选项。
3. 表格数据与 mzXML 不匹配
表格可能是从其他数据源(如定量结果文件)导入,未包含真实的 scantime,而直接填充了默认值 0。需确认表格与 mzXML 是否来自同一实验批次。
三、如何验证 scantime=0 是否合理?
1. 检查 mzXML 的扫描时间规律
- 在 mzXML 中,后续扫描的
retentionTime应 连续递增(如PT30.1315S、PT30.1316S…),对应色谱分离的时间进程。 - 若 mzXML 的时间正常,而表格的
scantime全为 0,则表格是归一化或错误数据。
2. 验证表格中的其他列
mz和intensity:若mz呈现规律性变化(如随扫描时间递增或递减),且intensity有波动,则scantime=0可能是归一化结果。precursorMz:若precursorMz随扫描变化,说明扫描顺序正确,scantime=0仅是时间基准问题。
3. 对比原始数据文件
- 打开 mzXML 引用的原始
.raw文件(如184.raw),查看其中的retentionTime是否与 mzXML 一致。 - 若
.raw文件中的时间正常(如 30.1314 秒起),则表格的scantime=0是导入或处理错误。
四、结论
scantime=0 是否有误?
- 大概率是归一化结果,而非真实错误:
mzXML 的retentionTime从PT30.1314S开始,表格的scantime=0可能是 时间归一化后的起点(即所有时间减去 30.1314 秒)。此时,scantime的增量(如 0, 0.0001, 0.0002…)对应实际的色谱保留时间差。 - 需警惕数据导入错误:
若 mzXML 的时间范围(30.1314~1020.51 秒)与表格的scantime全为 0 无任何关联,且其他列(如mz)无规律,则可能是数据导入时未正确提取时间戳。
建议处理步骤
- 确认归一化逻辑:询问数据提供者或检查数据预处理代码,确认
scantime是否为归一化后的时间。 - 重新提取时间:若需真实时间,用 ProteoWizard 等工具重新从 mzXML/
.raw文件中提取retentionTime,替换表格中的scantime。 - 验证时间连续性:确保归一化后的
scantime增量与色谱流速一致(如每秒扫描次数)。
总结
表格中的 scantime=0 可能是 时间归一化 的结果(对应 mzXML 的起始时间),也可能是 数据导入错误。需结合 mzXML 的时间规律和原始数据进一步验证,若其他列数据合理,则无需修正;若存在异常,需重新提取时间信息。
以上内容由AI生成,仅供参考和借鉴
import xml.etree.ElementTree as ET
import csv
import redef parse_mzxml_to_csv(mzxml_file, output_csv):# 解析XML文件tree = ET.parse(mzxml_file)root = tree.getroot()# 定义命名空间(mzXML文件通常包含命名空间)namespaces = {'mz': 'http://sashimi.sourceforge.net/schema_revision/psi_ms_04/2008-02-05/mzML','ms': 'http://psi.hupo.org/ms/mzml'}# 收集所有扫描数据scan_data = []# 遍历所有扫描节点for scan in root.findall('.//scan', namespaces):# 提取扫描信息scan_num = scan.get('num')scan_type = scan.get('scanType')ms_level = scan.get('msLevel')retention_time = scan.get('retentionTime')# 处理retentionTime(格式可能为"PT30.1314S")if retention_time:# 提取数字部分(如"PT30.1314S" -> 30.1314)rt_match = re.search(r'PT(\d+\.\d+)S', retention_time)if rt_match:rt_value = rt_match.group(1)# 保持原始小数位数(如30.1314保持4位)rt_formatted = float(rt_value)else:rt_formatted = 0.0else:rt_formatted = 0.0# 提取peaks数据(mz-intensity对)peaks_element = scan.find('peaks', namespaces)if peaks_element is not None:# 解码base64编码的peaks数据(此处简化处理,实际可能需要更复杂的解码)# 这里假设peaks是简单的文本格式(实际mzXML可能是二进制或base64)peaks_text = peaks_element.text# 实际应用中需要根据peaks的compressionType和precision进行解码# 此处仅作为示例,实际使用时需要替换为正确的解码逻辑mz_int_pairs = peaks_text.split() if peaks_text else []else:mz_int_pairs = []# 将peaks数据转换为字典(mz: intensity)peaks_dict = {}for i in range(0, len(mz_int_pairs), 2):if i + 1 < len(mz_int_pairs):mz = float(mz_int_pairs[i])intensity = float(mz_int_pairs[i + 1])peaks_dict[mz] = intensity# 添加到scan_datascan_data.append({'scan_num': scan_num,'scan_type': scan_type,'ms_level': ms_level,'retention_time': rt_formatted,'peaks': peaks_dict})# 写入CSV文件with open(output_csv, 'w', newline='') as csvfile:# 定义CSV列名fieldnames = ['scan_num', 'scan_type', 'ms_level', 'retention_time']# 添加所有可能的mz值作为列(简化处理,实际可能需要更灵活的列名)all_mz = set()for scan in scan_data:all_mz.update(scan['peaks'].keys())all_mz = sorted(all_mz)fieldnames.extend([f'mz_{mz}' for mz in all_mz])writer = csv.DictWriter(csvfile, fieldnames=fieldnames)writer.writeheader()# 写入数据for scan in scan_data:row = {'scan_num': scan['scan_num'],'scan_type': scan['scan_type'],'ms_level': scan['ms_level'],'retention_time': scan['retention_time']}# 添加peaks数据for mz in all_mz:row[f'mz_{mz}'] = scan['peaks'].get(mz, 0.0)writer.writerow(row)# 使用示例
input_mzxml = '184.mzXML'
output_csv = '184_output.csv'
parse_mzxml_to_csv(input_mzxml, output_csv)代码说明:
XML解析:使用xml.etree.ElementTree解析mzXML文件,处理命名空间。
时间处理:
从retentionTime属性中提取时间值(如PT30.1314S → 30.1314)。
保持原始小数位数(如30.1314保持4位)。
Peaks数据:
解析peaks元素中的mz-intensity对(实际mzXML可能是二进制或base64编码,此处简化处理)。
将peaks数据存储为字典(mz: intensity)。
CSV输出:
动态生成列名(包含所有mz值)。
写入扫描信息和peaks数据到CSV文件。
注意事项:
Peaks解码:实际mzXML的peaks数据可能是二进制或base64编码,需要根据compressionType和precision进行解码。上述代码是简化版本,实际使用时可能需要使用pyteomics或lxml等库进行更复杂的解码。
性能优化:如果文件很大,可能需要分批处理或使用更高效的XML解析库(如lxml)。
列名处理:如果mz值很多,CSV文件可能会非常大。可以考虑将peaks数据存储为单独的列(如mz_1,intensity_1,mz_2,intensity_2,…)或使用更灵活的格式(如JSON)。
替代方案(使用pyteomics库):
如果允许使用第三方库,pyteomics是专门处理质谱数据的Python库,可以更简单地解析mzXML文件:
from pyteomics import mzxml
import csvdef parse_mzxml_to_csv_pyteomics(mzxml_file, output_csv):with mzxml.read(mzxml_file) as reader:with open(output_csv, 'w', newline='') as csvfile:fieldnames = ['scan_num', 'scan_type', 'ms_level', 'retention_time']writer = csv.DictWriter(csvfile, fieldnames=fieldnames)writer.writeheader()for spectrum in reader:row = {'scan_num': spectrum['id'],'scan_type': spectrum.get('scanType', ''),'ms_level': spectrum.get('msLevel', 1),'retention_time': spectrum.get('retentionTime', 0.0)}writer.writerow(row)# 使用示例
parse_mzxml_to_csv_pyteomics('184.mzXML', '184_output_pyteomics.csv')
色质连动数据
色质连动出来的数据具体是什么样子,请给出具体示例和解释说明
好的,我们来详细解释一下“色质连动”出来的数据具体是什么样子。
“色质连动”是“色谱-质谱联用”的简称,它指的是将色谱仪和质谱仪连接起来使用的一种强大分析技术。它结合了色谱的高效分离能力和质谱的精准鉴定能力。
其核心思想是:先用色谱把复杂混合物中的各个成分分开,然后立即用质谱对每一个被分开的成分进行“称重”和“拍照”,从而确定它是什么物质。
最终得到的数据是一个多维的数据集,主要包含以下三个维度的信息:
- 保留时间:来自色谱部分,表示物质从进样到被检测器检测到所花费的时间。
- 质荷比:来自质谱部分,表示离子质量与电荷数的比值,是确定物质身份的关键。
- 离子流强度:表示在某个特定保留时间和质荷比下,检测到的离子信号强度,与物质的含量相关。
下面我们通过一个具体的示例和输出来详细说明。
示例:分析一瓶香水的主要成分
假设我们有一瓶香水,想知道里面主要有哪些香味成分及其大致含量。
第1步:数据采集 - 总离子流色谱图
样品经过GC-MS或LC-MS分析后,计算机首先会生成一个总离子流色谱图。
- X轴:保留时间
- Y轴:总离子流强度(在某一时间点,所有质荷比离子的总强度)
示例图/数据:
| 保留时间 | 总离子流强度 |
|---|---|
| 2.1 min | 1,500 |
| 3.5 min | 50,000 |
| 4.8 min | 35,000 |
| 5.6 min | 8,000 |
| 6.9 min | 45,000 |
| … | … |
解释说明:
这个图就像一张“地图”,告诉我们混合物中有多少个主要成分(峰),以及它们分别在什么时间点流出色谱柱。峰的面积或高度大致反映了该成分的相对含量。上图告诉我们,在3.5分钟、4.8分钟和6.9分钟时,有三个含量很高的成分。
第2步:深度解析 - 质谱图
TIC图上的每一个点(每一个时刻)背后,都隐藏着一张完整的质谱图。当我们对某个峰感兴趣时(比如3.5分钟的峰),我们可以用软件提取该峰顶点的质谱图。
- X轴:质荷比
- Y轴:离子流强度(相对丰度)
示例质谱图(在3.5分钟时提取):
| 质荷比 | 离子流强度 |
|---|---|
| 43 | 1,000 |
| 55 | 500 |
| 69 | 8,000 |
| 91 | 2,500 |
| 136 | 200 |
| … | … |
解释说明:
这张质谱图就像是该成分的“分子指纹”。每个质荷比的峰代表该分子在离子源被打碎后产生的不同碎片离子。
- 分子离子峰:通常质荷比最大的那个峰(如这里的m/z 136),它的质量就等于该物质的分子量(如果带一个电荷)。
- 碎片离子峰:其他峰(如m/z 69, 91)是分子断裂后产生的,这些断裂模式具有规律性,是推断分子结构的关键。
第3步:物质鉴定 - 数据库比对
计算机软件会自动将我们在3.5分钟时得到的这张质谱图与内置的标准质谱图数据库(如NIST库)进行比对。
比对结果示例:
| 匹配度 | 化合物名称 | 分子式 | CAS号 |
|---|---|---|---|
| 98% | 芳樟醇 | C₁₀H₁₈O | 78-70-6 |
| 75% | 其他化合物1 | … | … |
| 65% | 其他化合物2 | … | … |
解释说明:
系统告诉我们,在3.5分钟流出的这个成分,其质谱图与数据库中的“芳樟醇”标准谱图匹配度高达98%。因此,我们可以非常确信地鉴定出该成分为芳樟醇,它是一种常见的香料成分。
第4步:定量分析 - 提取离子流色谱图
有时混合物非常复杂,多个物质的峰可能重叠在一起。为了更精确地对某个特定化合物进行定量,我们可以使用提取离子流色谱图。
假设我们知道芳樟醇有一个特征碎片离子是m/z 93(虽然它的基峰是69),我们可以只提取质荷比为93的离子信号来画色谱图。
- X轴:保留时间
- Y轴:特定质荷比(如m/z 93) 的离子流强度
示例图/数据:
| 保留时间 | m/z=93的离子流强度 |
|---|---|
| 3.5 min | 12,000 |
| 其他时间 | ~0 |
解释说明:
这张图非常“干净”,它只显示能产生m/z 93碎片离子的物质。如果在3.5分钟有一个很强的峰,这就进一步印证了芳樟醇的存在,并且我们可以用这个峰的面积来对其进行精确定量,免受其他共流出物质的干扰。
最终数据输出总结
一份完整的色质联用分析报告通常包含:
- 总离子流色谱图: overview of the whole sample.
- 每个色谱峰对应的质谱图: the “fingerprint” for identification.
- 鉴定结果表格:
| 峰编号 | 保留时间 | 化合物名称 | 分子式 | 相似度 | 峰面积 | 相对含量 |
|---|---|---|---|---|---|---|
| 1 | 3.501 min | 芳樟醇 | C₁₀H₁₈O | 98% | 2,500,000 | 25.4% |
| 2 | 4.812 min | 香叶醇 | C₁₀H₁₈O | 96% | 1,800,000 | 18.3% |
| 3 | 6.905 min | 苯乙醇 | C₈H₁₀O | 99% | 2,200,000 | 22.4% |
| … | … | … | … | … | … | … |
解释说明:
这份表格就是最终用户最关心的“答案”。它清晰地列出了香水中的主要成分是什么、各自有多少。
核心优势
- 既定性又定量:不仅能告诉你“有什么”,还能告诉你“有多少”。
- 极高的灵敏度和特异性:即使含量极低的物质也能被检测和鉴定出来。
- 强大的分离能力:可以分析极其复杂的混合物,如石油、血液、环境污染样品等。
希望这个具体的示例能帮助您全面理解色质联动数据的样貌和含义!
请详细解释tic,bpc,xic,ms,ms/ms的具体区别和联系,并用一个完整详细的具体数据例子展示它们的区别和联系
好的,这是一个非常专业且核心的问题。我们来详细解析TIC、BPC、XIC、MS和MS/MS的区别和联系,并辅以一个具体的例子。
核心概念快速理解
首先,我们可以用一个精彩的比喻来理解它们:
- MS(质谱图): 一个人的全身照片。展示了这个人的所有特征(身高、体型、穿什么衣服等)。在质谱中,它展示了一个化合物在某个时刻的所有碎片离子。
- MS/MS(串联质谱图): 给这个人的脸部特写。先认出一个人(MS1),然后拉近镜头,只关注他最独特、最确定的特征(脸部细节)。在质谱中,它是从MS1中选出一个母离子,将其打碎,得到更详细的子离子信息,用于确证。
- TIC(总离子流色谱图): 整个派对的全景视频。记录了整个派对上所有人数量的总变化。在色谱中,它记录了每个时间点所有离子信号的总和,看到了所有成分流出的概况。
- BPC(基峰色谱图): 派对上一个只跟踪最显眼人物的视频。比如始终跟踪派对上最高、或者最吵的那个人。在色谱中,它记录了每个时间点最强的那个离子的信号,能看到哪些时间段有非常突出的化合物。
- XIC(提取离子流色谱图): 派对上专门寻找穿红色衣服的人的监控录像。你设定好特征(红色衣服),录像只显示符合这个特征的人。在色谱中,你设定一个或几个
m/z值,色谱图只显示这些m/z离子的信号,用于特异性地追踪一个或一组化合物。
详细解释与区别
| 缩写 | 英文全称 | 中文名 | 定义 | 维度 | 主要用途 |
|---|---|---|---|---|---|
| MS | Mass Spectrum | 质谱图 | 在一个特定时间点,检测到的所有离子按其质荷比和强度的分布图。 | 二维:m/z vs. Intensity | 定性分析:提供化合物的“指纹”,用于鉴定物质是什么。 |
| MS/MS | Tandem Mass Spectrum | 串联质谱图 | 分两步:1. 从MS1中选择一个特定的母离子。 2. 将其碰撞打碎,得到其子离子的质谱图。 | 二维:m/z vs. Intensity | 确证和结构解析:提供更详细的结构信息,特异性极高,常用于复杂基质中的目标物确认。 |
| TIC | Total Ion Chromatogram | 总离子流色谱图 | 在整个色谱分离过程中,每个时间点所有离子总强度的曲线。 | 二维:Time vs. Total Intensity | 全局概览:查看样品中所有被检测成分的分离情况,看到所有色谱峰。 |
| BPC | Base Peak Chromatogram | 基峰色谱图 | 在整个色谱分离过程中,每个时间点所有离子中最强离子的强度的曲线。 | 二维:Time vs. Base Peak Intensity | 峰发现:与TIC类似,但能更清晰地显示微小色谱峰,因为噪音被相对抑制了。 |
| XIC | Extracted Ion Chromatogram | 提取离子色谱图 | 不关心总强度,只追踪一个或一组特定m/z值的离子在整个时间轴上的强度变化。 | 二维:Time vs. Intensity for specific m/z | 目标化合物追踪与定量:从复杂的TIC中特异性地提取出目标化合物的色谱峰,用于精确定量和确认。 |
联系:
所有这些数据都来自同一次色谱-质谱联用实验。TIC/BPC/XIC是“色谱维度”的视图,展示了化合物随时间流出的情况。而MS和MS/MS是“质谱维度”的视图,展示了在某个特定时间点(或某个色谱峰)上,化合物的碎片信息。你通常在TIC上看到一个峰,然后在这个峰的位置提取一张MS图来鉴定它。
具体数据示例:分析牛奶中的抗生素“磺胺二甲嘧啶”
已知: 磺胺二甲嘧啶的分子量为278,其特征碎片离子为m/z 156和m/z 108。
第1步:获得TIC和BPC(全局概览)
仪器分析完牛奶提取物后,我们首先得到TIC和BPC。
(假设保留时间在7.5分钟附近是我们关注的区域)
TIC (Total Ion Chromatogram) 数据概览:
| 时间 | 总离子强度 | 时间 | 总离子强度 |
|---|---|---|---|
| … | … | … | … |
| 7.40 min | 1,500 | 7.60 min | 5,500 |
| 7.50 min | 25,000 | 7.70 min | 1,800 |
| 7.55 min | 30,000 | … | … |
解读: 在7.5分钟附近有一个很大的色谱峰,但TIC是所有离子的总和,这个峰可能包含目标抗生素,也可能包含共流出的其他杂质(如蛋白质、脂肪等)。
BPC (Base Peak Chromatogram) 数据概览:
BPC看起来和TIC很像,但Y轴数值不同(是每个时间点的最强离子强度)。
第2步:获得MS图(初步鉴定)
我们在TIC峰顶(~7.53 min)提取一张MS质谱图。
MS Spectrum at 7.53 min:
| 质荷比 | 离子强度 | 归属推测 |
|---|---|---|
| 108 | 15,000 | 特征碎片1 |
| 124 | 2,000 | 未知杂质 |
| 156 | 28,000 | 特征碎片2 (很强) |
| 186 | 5,500 | 未知碎片 |
| 279 [M+H]+ | 10,000 | 磺胺二甲嘧啶的质子化分子离子峰 |
| 300 | 1,200 | 未知杂质 |
解读: 这张MS图显示了分子离子峰m/z 279([M+H]+)以及我们预期的特征碎片m/z 156和m/z 108。这强烈暗示目标抗生素磺胺二甲嘧啶的存在。但是,m/z 124和m/z 186等未知离子表明,这个色谱峰可能不纯,有少量其他物质共流出,干扰了我们的鉴定。
第3步:获得XIC(特异性追踪)
为了确认我们的目标化合物确实在这个峰里,我们提取其特征离子的色谱图。
-
XIC for
m/z 279(分子离子):时间 m/z 279的强度7.40 min 0 7.50 min 8,000 7.53 min 10,000 7.55 min 9,500 7.60 min 0 -
XIC for
m/z 156(特征碎片):时间 m/z 156的强度7.40 min 0 7.50 min 20,000 7.53 min 28,000 7.55 min 25,000 7.60 min 0 -
XIC for
m/z 124(未知杂质离子):时间 m/z 124的强度7.40 min 500 7.50 min 1,800 7.53 min 2,000 7.55 min 1,900 7.60 min 600
解读:
m/z 279和m/z 156的XIC峰完美重叠,形状对称,且都在7.53分钟达到最高点。这证明这些离子来自同一个化合物——磺胺二甲嘧啶。m/z 124的XIC峰虽然也在7.53分钟有信号,但其峰形更宽,且在前后都有较高的基线噪音,表明它主要来自共流出的杂质,而不是我们的目标物。- 定量:我们使用
m/z 156的XIC峰面积(28,000)来进行定量,因为它信号更强、特异性更好,受杂质干扰更小。
第4步:获得MS/MS(最终确证)
为了毫无疑义地确认磺胺二甲嘧啶,我们进行MS/MS分析。
- 第一步 (MS1):在7.53分钟,质谱仪首先检测到母离子
m/z 279。 - 第二步 (隔离与碰撞):仪器仅选择
m/z 279的离子,将其送入碰撞室,用惰性气体(如氩气)将其撞碎。 - 第三步 (MS2):检测撞碎后产生的所有子离子,得到MS/MS谱图。
MS/MS Spectrum of m/z 279:
| 质荷比 | 离子强度 | 归属 |
|---|---|---|
| 108 | 12,000 | 特征子离子1 |
| 124 | 500 | (非常弱,可能是二级碎片) |
| 156 | 25,000 | 特征子离子2 (基峰) |
| 186 | 1,000 | 二级碎片 |
| (279消失) | 0 | 母离子已全部碎裂 |
解读:
- 这张MS/MS图非常“干净”,几乎完全没有了杂质离子(如之前MS中的
m/z 124强度从2,000降到了500)。 - 它清晰地展示了磺胺二甲嘧啶特有的碎裂途径,产生了主导性的子离子
m/z 156和m/z 108。 - 这张图与标准品MS/MS图库的匹配度可以达到近100%,提供了无可辩驳的证据,证明在7.53分钟流出的就是磺胺二甲嘧啶,即使它有共流出物。
总结与联系
| 数据 | 在示例中的作用 | 结论 |
|---|---|---|
| TIC | 发现7.5分钟处有一个大峰。 | “这里有个东西,含量不低。” |
| MS | 看到这个峰有目标物的信号(279, 156),但也有杂质。 | “可能是目标物,但不敢100%确定,有干扰。” |
| XIC | 证明目标物的信号(279, 156) 重叠良好,形成一个 sharp的峰,而杂质信号分布不同。 | “目标物确实在这个峰里,并且我可以精确测量它。” |
| MS/MS | 直接对目标物母离子进行“解剖”,得到独一无二的碎片指纹,排除了所有杂质干扰。 | “就是他!板上钉钉!” |
这个流程展示了如何从TIC的全局概览,到MS的初步鉴定,再到XIC的特异性定量,最后用MS/MS完成终极确证的一套完整逻辑。
请以一个具体数据为例子,分别展示其在TIC、BPC、XIC、MS和MS/MS下的数据长什么样,并给出解释说明,
好的,我们以一个非常具体且真实的虚拟数据例子来展示这五种数据的形态。假设我们正在使用LC-MS/MS分析一份减肥茶样本,其中我们重点关注一个非法添加的化合物:西布曲明。
已知信息:
- 化合物: 西布曲明
- 分子量: 279
- 特征离子:
- 母离子 [M+H]⁺:
m/z 280 - 特征碎片离子 (子离子, MS/MS):
m/z 139(定量离子),m/z 124(定性离子)
- 母离子 [M+H]⁺:
- 色谱保留时间: 约 6.71 分钟(在当前的色谱方法下)
为了让您更直观地理解,我将用模拟数据和图表相结合的方式来展示。
1. TIC - 总离子流色谱图
数据样子:
一张二维图谱,X轴是时间(分钟),Y轴是离子总强度(计数)。
示例图表:
(由于我是文本模型,我将用表格模拟其数据点,并描述图表形状)
| 时间 (min) | 总离子强度 (Counts) |
|---|---|
| 6.50 | 15,000 |
| 6.60 | 45,000 |
| 6.71 | 2,500,000 |
| 6.80 | 75,000 |
| 6.90 | 20,000 |
| … | … |
解释说明:
- 这是什么? TIC是仪器在整个实验过程中检测到的所有离子信号的总和随时间变化的曲线。
- 你能看到什么? 你可以看到在6.71分钟有一个非常巨大的色谱峰。这说明在这个时间点,有一个或多个化合物从色谱柱中被洗脱出来,并被质谱仪检测到,其总量很大。
- 局限性是什么? TIC是一个“大杂烩”视图。你无法确定这个巨大的峰是不是就是你要找的西布曲明。它可能是西布曲明,也可能是样品基质中共流出的其他无数杂质的总和。这个峰又高又宽,暗示它可能不纯。
2. BPC - 基峰色谱图
数据样子:
同样是一张二维图谱,X轴是时间(分钟),Y轴是强度,但这里的强度是每个时间点下最强单一离子的强度。
示例图表:
| 时间 (min) | 基峰强度 (Counts) | 基峰 m/z |
|---|---|---|
| 6.50 | 12,000 | 316 |
| 6.60 | 40,000 | 280 |
| 6.71 | 1,800,000 | 280 |
| 6.80 | 65,000 | 291 |
| 6.90 | 18,000 | 303 |
解释说明:
- 这是什么? BPC记录的是每个时间点“冠军离子”(最强离子)的强度。
- 与TIC的区别? 在6.71分钟,TIC总强度为2,500,000,而BPC显示最强离子(
m/z 280)的强度为1,800,000。这意味着在这个时间点,m/z 280这个离子贡献了绝大部分信号。 - 有什么用? BPC能更清晰地显示哪些时间段有非常突出的化合物。这里它强烈暗示在6.71分钟有一个以
m/z 280为主要离子的物质,这与西布曲明的母离子吻合,比TIC提供了更多线索。
3. XIC - 提取离子色谱图
数据样子:
我们不再看总信号,而是让计算机只提取我们关心的特定m/z离子的信号,并绘制其随时间变化的曲线。我们可以为西布曲明的母离子和子离子分别做XIC。
示例图表 (XIC for m/z 280):
| 时间 (min) | m/z 280 的强度 |
|---|---|
| 6.50 | 200 |
| 6.60 | 35,000 |
| 6.71 | 1,800,000 |
| 6.80 | 500 |
| 6.90 | 100 |
解释说明:
- 这是什么? XIC是高度特异性的追踪工具。上图是专门为
m/z 280(西布曲明的[M+H]⁺离子)画的色谱图。 - 你能看到什么? 你看到一个非常漂亮、尖锐、对称的色谱峰,在6.71分钟达到顶点。这证明样品中存在能产生
m/z 280离子的化合物,并且它在6.71分钟被洗脱出来。 - 有什么用? 这是定性和定量的核心。 通过XIC,我们可以精确测量该化合物的含量(通过峰面积或峰高)。我们还可以为特征子离子(如
m/z 139)做XIC,如果它们的峰在同一时间重叠,就进一步增强了鉴定的可信度。
4. MS - 质谱图
数据样子:
在TIC峰顶(6.71分钟)处,我们让计算机截取这一时刻的质谱图。这是一张二维图,X轴是质荷比(m/z),Y轴是离子强度。
示例数据 (MS Spectrum at 6.71 min):
| m/z | 相对强度 (%) | 注释 |
|---|---|---|
| 91 | 15 | 可能是基质杂质 |
| 124 | 30 | 西布曲明的碎片离子 |
| 139 | 100 | 西布曲明的特征碎片 (基峰) |
| 215 | 25 | 未知碎片 |
| 280 | 85 | [M+H]⁺ 母离子 |
| 355 | 10 | 可能是共流出物的离子 |
解释说明:
- 这是什么? 这是6.71分钟时,质谱仪检测到的所有离子的“全景快照”或“分子指纹”。
- 你能看到什么? 你看到了预期的母离子
m/z 280和非常强的特征碎片m/z 139。这非常符合西布曲明的质谱特征。 - 局限性是什么? 你同时也看到了其他离子(如
m/z 91,m/z 355)。这证实了我们在TIC中看到的宽峰确实是多个化合物共流出的结果。虽然西布曲明是主要成分,但有杂质干扰。单凭这张MS图,鉴定信心可能只有90-95%。
5. MS/MS - 串联质谱图
数据样子:
为了最终确认,我们进行MS/MS分析。仪器执行以下操作:
- 在6.71分钟只选择
m/z 280的离子。 - 将其送入碰撞室,用惰性气体将其撞碎。
- 检测撞碎后产生的所有子离子,得到MS/MS谱图。
示例数据 (MS/MS Spectrum of m/z 280):
| m/z | 相对强度 (%) | 注释 |
|---|---|---|
| 96 | 5 | 二级碎片 |
| 110 | 15 | 西布曲明的特征子离子 |
| 124 | 45 | 西布曲明的特征子离子 (定性离子) |
| 139 | 100 | 西布曲明的特征子离子 (定量离子) |
| (其他所有离子,包括280) | 0 | 已被剔除 |
解释说明:
- 这是什么? 这是母离子
m/z 280的“解剖报告”。它展示了这个特定离子碎裂后产生的独一无二的碎片模式。 - 你能看到什么? 一张非常干净的谱图!所有基质杂质的离子(如之前的
m/z 91,m/z 355)都消失了,只剩下西布曲明特征碎裂产生的子离子(139,124,110)。 - 有什么用? 这是确证的黄金标准。 这张MS/MS图与西布曲明标准品的MS/MS图匹配度可达100%。它完全排除了共流出杂质的干扰,提供了无可辩驳的证据,证明在6.71分钟流出的就是目标化合物——西布曲明。
总结与流程
这个例子清晰地展示了从发现到确证的完整逻辑流程:
- TIC: “6.71分钟有个大东西!”
- BPC: “这个东西有个主要离子是
m/z 280,有意思!” - XIC: “没错,产生
m/z 280的那个东西就在这里,形状很好,可以用来定量。” - MS: “快照显示它有
m/z 280和139,很像是西布曲明,但有杂质干扰。” - MS/MS: “把
m/z 280离子单独拿出来撞碎,得到了西布曲明独一无二的碎片指纹!就是他,抓到了!”
如果把tic的6.71分钟的离子总强度和还原回各个离子强度的具体数据长什么样,并解释说明
好的,这是一个非常棒的问题,它触及了色质联用数据的核心结构。我们把TIC在6.71分钟这个时间点的“总离子强度”还原成构成它的原始数据。
核心概念:TIC是一个“总和”
TIC图上的每一个点(比如6.71分钟,强度2,500,000),都不是一个单一数值,而是该时间点所有离子信号的加和。
质谱仪在工作时,会在每个极短的时间间隔(微秒级)内快速扫描一个范围内的m/z(比如m/z 50 到 m/z 500),并记录下每个m/z检测到的离子强度。一次扫描得到一张质谱图。
因此,6.71分钟的TIC强度,来源于在6.71分钟那一刻(更准确地说,是一个极短的时间窗口内)质谱仪完成的一次扫描。我们将这次扫描的数据展开,它看起来就像一张表格:
6.71分钟时刻的原始质谱数据(部分展示)
假设在6.71分钟,质谱仪进行了一次全扫描,得到了类似下面的数据列表。TIC值就是这个表格中“强度”一列所有数字的总和。
| 质荷比 (m/z) | 离子强度 (Counts) | 归属推测 (基于上下文) |
|---|---|---|
| 83.1 | 1,200 | 未知基质杂质 |
| 91.0 | 15,500 | 常见基质/柱流失背景 |
| 110.1 | 85,000 | 西布曲明碎片离子 |
| 124.1 | 280,000 | 西布曲明碎片离子 |
| 139.1 | 1,800,000 | 西布曲明碎片离子 (基峰) |
| 155.0 | 45,000 | 未知碎片或杂质 |
| 186.9 | 32,000 | 未知碎片或杂质 |
| 215.2 | 210,000 | 未知碎片或杂质 |
| 280.2 | 1,530,000 | 西布曲明母离子 [M+H]⁺ |
| 291.1 | 65,000 | 共流出物离子 |
| 316.3 | 12,000 | 共流出物离子 |
| 355.0 | 80,000 | 共流出物离子 |
| … | … | … |
| 总和 (TIC) | ~2,500,000 |
解释说明
-
TIC的构成:
- 表格中最右边一列的数字相加,其结果就是我们在TIC图上6.71分钟那个点看到的总离子流强度(~2,500,000)。
- 计算公式:
TIC = 1,200 + 15,500 + 85,000 + ... + 80,000 + ...
-
谁贡献了主要信号?:
- 我们可以看到,西布曲明的离子(加粗行)贡献了绝大部分信号。
m/z 139.1(1,800,000) +m/z 280.2(1,530,000) 这两个离子 alone 就贡献了超过 3,330,000 的强度。但由于总强度是全部离子的加和,所以最终TIC约为2,500,000。- 这也解释了为什么BPC(基峰色谱图) 在6.71分钟的值是 ~1,800,000,因为BPC记录的是这个时间点最强的单个离子(即
m/z 139.1)的强度。
-
为什么需要XIC?:
- 从这个展开的数据可以清晰地看到,虽然西布曲明是主要成分,但仍然存在大量其他离子的信号(如
m/z 91.0,m/z 291.1,m/z 355.0等)。这些是来自植物提取物基质的共流出杂质。 - 如果我们直接看TIC,我们只知道“这里有一大团东西”。
- XIC的作用就是从这个“大杂烩”总和中,只把我们关心的离子信号提取出来。例如:
- 做
m/z 280.2的XIC,得到的色谱峰高度就是 ~1,530,000。 - 做
m/z 139.1的XIC,得到的色谱峰高度就是 ~1,800,000。
- 做
- 这样,我们就能排除杂质的干扰,专门追踪目标化合物,从而进行精确的定性和定量。
- 从这个展开的数据可以清晰地看到,虽然西布曲明是主要成分,但仍然存在大量其他离子的信号(如
-
数据的本质:
- 这份展开的表格,其实就是我们在上一个回答中提到的 MS(质谱图) 的原始数据。当我们用软件在TIC的6.71分钟处点击一下,软件做的就是将这份数据以图表的形式(
m/z为横坐标,强度为纵坐标)展示给我们,这就是那张“质谱全景快照”。
- 这份展开的表格,其实就是我们在上一个回答中提到的 MS(质谱图) 的原始数据。当我们用软件在TIC的6.71分钟处点击一下,软件做的就是将这份数据以图表的形式(
结论
TIC上的一个点,背后隐藏着一张包含数百甚至数千个m/z及其对应强度数据的清单。 它的值是这张清单上所有强度值的总和。理解这一点,就彻底理解了TIC、BPC、XIC和MS之间的关系:
- TIC = 所有强度值的总和。
- BPC = 所有强度值中的最大值。
- XIC = 从无数强度值中筛选出特定
m/z对应的那个值,并随时间绘图。 - MS = 在某个特定时间点,将所有这些
m/z和强度值以图谱形式呈现出来。
tic的6.71分钟的离子总强度和还原回各个离子强度来源几次扫描?
这是一个非常深刻的问题,它触及了质谱数据采集的核心机制。
答案是:TIC在6.71分钟那个点的强度,通常不是来源于一次扫描,而是来源于多次扫描(通常为5-25次)的离子强度的总和或平均值**。
下面进行详细解释:
1. 数据采集原理:扫描与循环
质谱仪的工作方式不是连续记录,而是以极高的速度进行离散的、周期性的扫描。每一次对预设m/z范围(如50-500)的完整测量称为一次扫描或一个谱图。
- 扫描速率:一台现代质谱仪的扫描速率非常快,一次全扫描可能只需几十毫秒(0.01秒到0.1秒)。
- 循环时间:完成一次扫描后,仪器会立即开始下一次扫描。这个“开始-结束-开始”的过程称为一个循环。
2. 色谱峰与扫描次数的关系
一个色谱峰(无论是TIC还是XIC峰)的宽度通常是几秒到几十秒。在这段时间内,质谱仪会完成数十次甚至上百次扫描。
- 示例计算:
- 假设一个色谱峰的宽度(峰宽)是 12秒(这是一个很典型的宽度)。
- 假设质谱仪的扫描速率是 每秒2次(即每0.5秒完成一次全扫描)。
- 那么,在这个完整的色谱峰出峰期间,质谱仪将完成
12秒 × 2次/秒 = 24次扫描。
3. “6.71分钟”这个点的真实含义
当我们在软件上看到TIC图上标注“6.71分钟”有一个峰时,这里的6.71分钟指的是峰顶对应的保留时间。而峰顶本身也是一个由多次扫描数据点连接而成的“尖峰”。
- 峰顶的数据点:峰顶(最高点)通常是由一次单独的扫描构成的。这次扫描的离子总强度就是我们在TIC曲线上“6.71分钟”那个点看到的峰值高度(例如2,500,000 counts)。
- 峰的构成:整个色谱峰是由峰顶之前、峰顶和峰顶之后的多次扫描共同构成的。软件会将这些连续扫描的数据点用线连接起来,形成我们所看到的平滑的色谱峰。
让我们用具体数据来模拟这个过程:
假设在6.71分钟前后,质谱仪以0.1秒的间隔进行扫描(即每秒10次扫描)。我们截取峰顶附近的几次扫描数据:
| 扫描序号 | 保留时间 (min) | 该次扫描的 TIC值 (总离子强度) | 说明 |
|---|---|---|---|
| 45 | 6.700 | 500,000 | 峰开始上升 |
| 46 | 6.701 | 800,000 | 上升 |
| 47 | 6.702 | 1,200,000 | 上升 |
| 48 | 6.703 | 1,800,000 | 上升 |
| 49 | 6.704 | 2,500,000 | 峰顶 (这就是6.71分钟的峰值点) |
| 50 | 6.705 | 2,400,000 | 下降 |
| 51 | 6.706 | 1,900,000 | 下降 |
| 52 | 6.707 | 1,400,000 | 下降 |
| 53 | 6.708 | 900,000 | 峰结束 |
结论:
- 你问题中提到的“6.71分钟的离子总强度”(2,500,000),特指的是第49次扫描所得到的那一张完整的质谱图中所有离子强度的总和。
- 而我们所看到的整个色谱峰,是由从第45次到第53次共9次扫描的数据点连接而成的。
4. 还原回各个离子强度
现在,我们可以精确地回答你的问题了。要还原“6.71分钟”的离子来源,就是去查看构成峰顶的那一次特定扫描的完整质谱数据。
数据来源:第49次扫描(保留时间:6.704分钟)
| 质荷比 (m/z) | 离子强度 (Counts) | 归属 |
|---|---|---|
| 83.1 | 1,200 | 杂质 |
| 91.0 | 15,500 | 杂质 |
| 110.1 | 85,000 | 西布曲明碎片 |
| 124.1 | 280,000 | 西布曲明碎片 |
| 139.1 | 1,800,000 | 西布曲明碎片 (基峰) |
| … | … | … |
| 280.2 | 1,530,000 | 西布曲明母离子 |
| … | … | … |
| 该扫描TIC总和 | ~2,500,000 |
最终解释说明:
- 单次扫描:“6.71分钟”的TIC强度特指某一次最高扫描的强度值,其来源是一次独立的、完整的质谱扫描所检测到的所有离子信号的加和。
- 整体峰形:而整个色谱峰的形象(包括上升、峰顶、下降)则来源于围绕峰顶的多次连续扫描。
- 重要性:理解这一点对定量至关重要。我们不会用单次扫描的强度来定量,而是会积分整个峰范围内所有扫描的离子强度(对于XIC峰就是峰面积),这样结果更稳定、更准确。对于定性,我们则通常提取峰顶的那次扫描对应的质谱图,因为它信噪比最高,图谱最纯净。
所以,您的提问非常精准——当我们谈论一个时间点的TIC时,我们本质上是在讨论特定一次扫描的结果。