生物信息学深度学习模型比较与学习框架
说起来是框架,其实是文献阅读,或者是模型比较时,一个逻辑清晰的结构化的思考框架:
主要是从宏观问题定义到微观技术细节,再到宏观评估与应用。
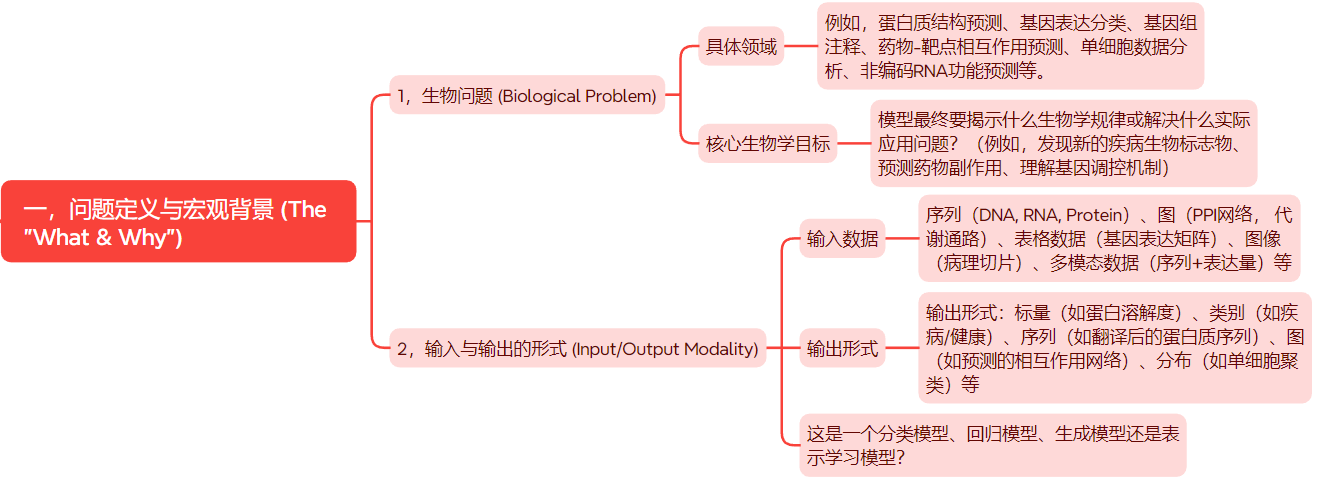
一,问题定义与宏观背景 (The “What & Why”)
第一层:问题定义与宏观背景 (The “What & Why”)
在深入模型之前,必须先理解它要解决的核心问题。
- 生物问题 (Biological Problem):
- 具体领域:例如,蛋白质结构预测、基因表达分类、基因组注释、药物-靶点相互作用预测、单细胞数据分析、非编码RNA功能预测等。
- 核心生物学目标:模型最终要揭示什么生物学规律或解决什么实际应用问题?(例如,发现新的疾病生物标志物、预测药物副作用、理解基因调控机制)。
- 输入与输出的形式 (Input/Output Modality):
- 输入数据:序列(DNA, RNA, Protein)、图(PPI网络, 代谢通路)、表格数据(基因表达矩阵)、图像(病理切片)、多模态数据(序列+表达量)等。
- 输出形式:标量(如蛋白溶解度)、类别(如疾病/健康)、序列(如翻译后的蛋白质序列)、图(如预测的相互作用网络)、分布(如单细胞聚类)等。
- 这是一个分类模型、回归模型、生成模型还是表示学习模型?
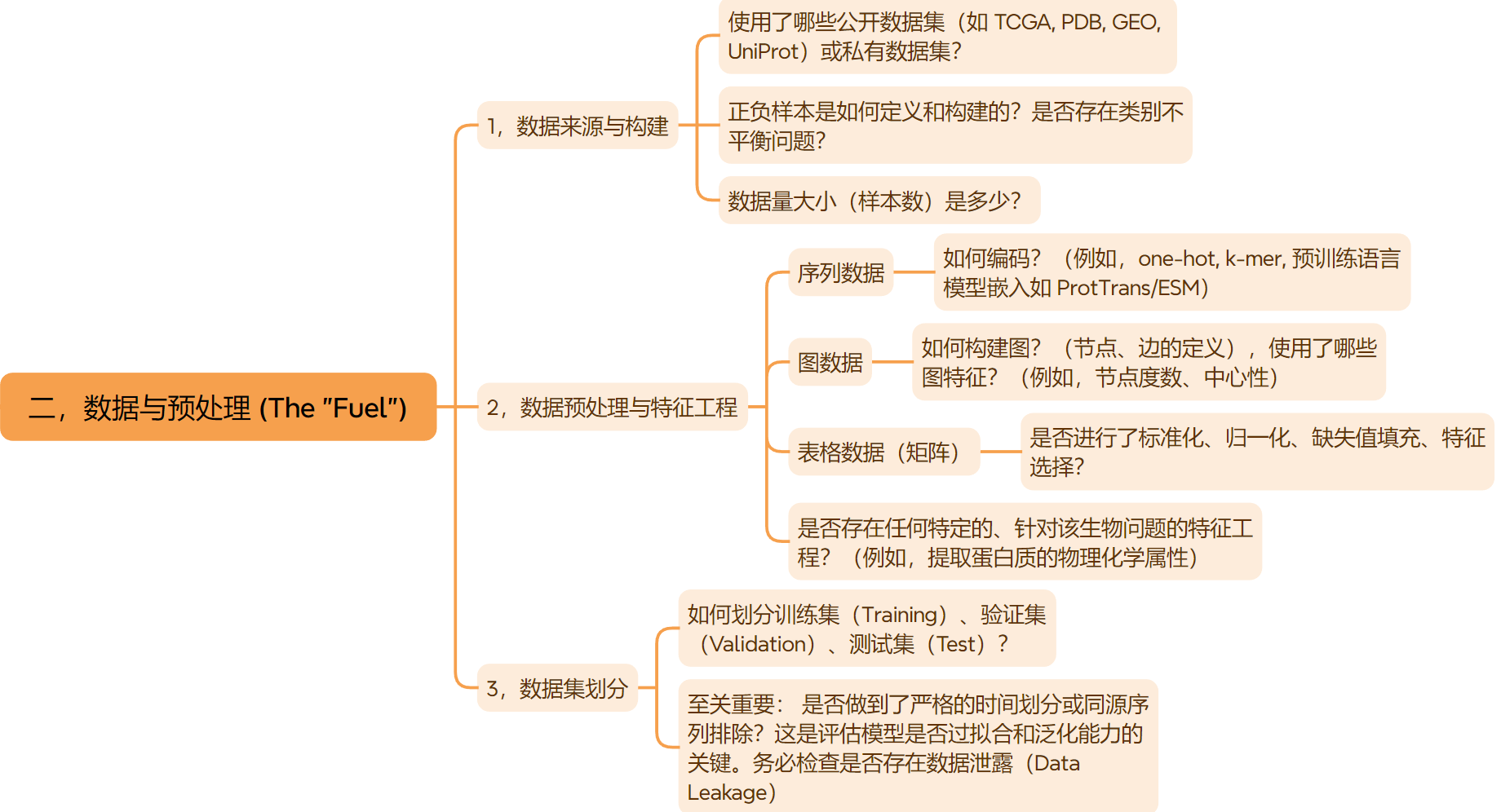
二,数据与预处理 (The “Fuel”)
第二层:数据与预处理 (The “Fuel”)
数据是模型的基石,在生物信息学中尤其重要,因为数据往往具有噪声高、不平衡、维度高等特点。
- 数据来源与构建:
- 使用了哪些公开数据集(如 TCGA, PDB, GEO, UniProt)或私有数据集?
- 正负样本是如何定义和构建的?是否存在类别不平衡问题?
- 数据量大小(样本数)是多少?
- 数据预处理与特征工程:
- 序列数据: 如何编码?(例如,one-hot, k-mer, 预训练语言模型嵌入如 ProtTrans/ESM)。
- 图数据: 如何构建图?(节点、边的定义),使用了哪些图特征?(例如,节点度数、中心性)。
- 表格数据: 是否进行了标准化、归一化、缺失值填充、特征选择?
- 是否存在任何特定的、针对该生物问题的特征工程?(例如,提取蛋白质的物理化学属性)。
- 数据集划分:
- 如何划分训练集(Training)、验证集(Validation)、测试集(Test)?
- 至关重要: 是否做到了严格的时间划分或同源序列排除?这是评估模型是否过拟合和泛化能力的关键。务必检查是否存在数据泄露(Data Leakage)。
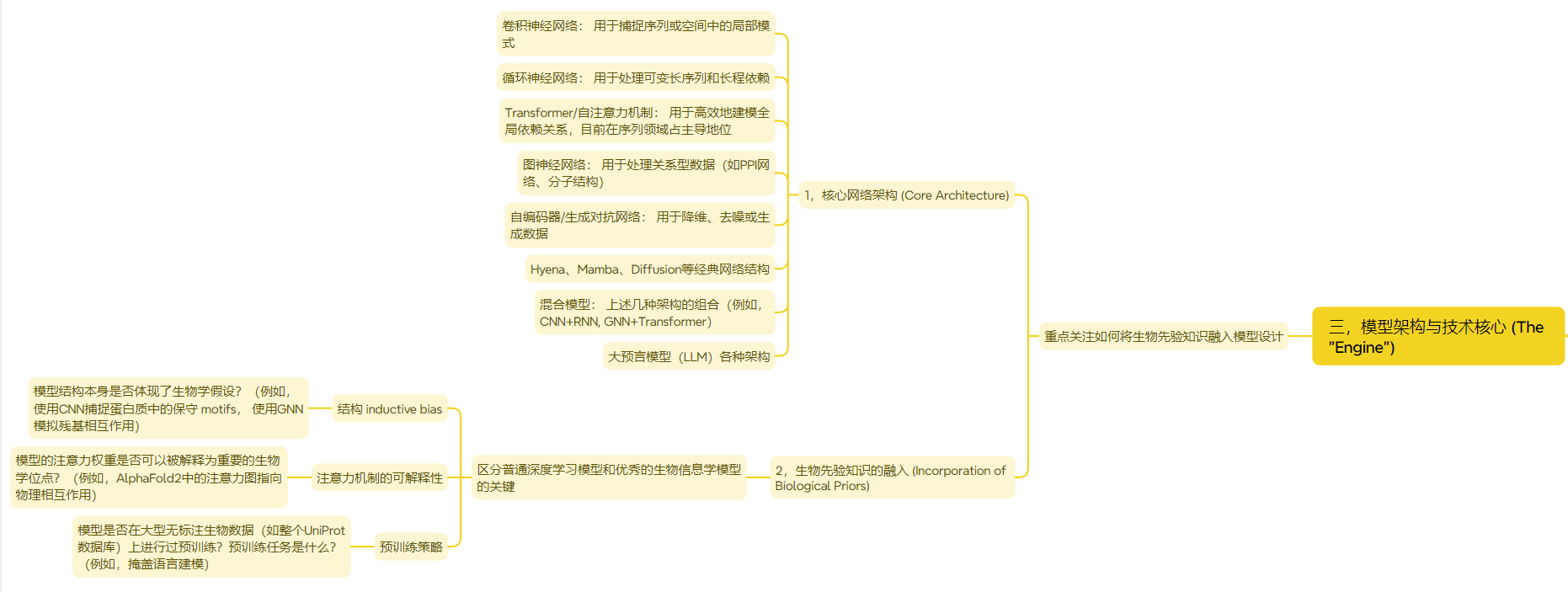
三,模型架构与技术核心 (The “Engine”)
第三层:模型架构与技术核心 (The “Engine”)
这是模型比较的核心,重点关注如何将生物先验知识融入模型设计。
- 核心网络架构 (Core Architecture):
- 卷积神经网络: 用于捕捉序列或空间中的局部模式。
- 循环神经网络: 用于处理可变长序列和长程依赖。
- Transformer/自注意力机制: 用于高效地建模全局依赖关系,目前在序列领域占主导地位。
- 图神经网络: 用于处理关系型数据(如PPI网络、分子结构)。
- 自编码器/生成对抗网络: 用于降维、去噪或生成数据。
- 混合模型: 上述几种架构的组合(例如,CNN+RNN, GNN+Transformer)。
- 生物先验知识的融入 (Incorporation of Biological Priors):
- 这是区分普通深度学习模型和优秀的生物信息学模型的关键!
- 结构 inductive bias: 模型结构本身是否体现了生物学假设?(例如,使用CNN捕捉蛋白质中的保守 motifs, 使用GNN模拟残基相互作用)。
- 注意力机制的可解释性: 模型的注意力权重是否可以被解释为重要的生物学位点?(例如,AlphaFold2中的注意力图指向物理相互作用)。
- 预训练策略: 模型是否在大型无标注生物数据(如整个UniProt数据库)上进行过预训练?预训练任务是什么?(例如,掩盖语言建模)。
四,训练与优化 (The “Tuning”)

第四层:训练与优化 (The “Tuning”)
- 损失函数 (Loss Function):
- 标准损失函数(如交叉熵、均方误差)还是为特定任务设计的自定义损失函数?(例如,处理不平衡数据的focal loss, 对比学习中的InfoNCE loss)。
- 优化策略 (Optimization):
- 使用了什么优化器(Adam, SGD)?学习率是多少?是否有学习率调度策略?
- 正则化与防止过拟合:
- 使用了哪些技术?(例如,Dropout, Weight Decay, Early Stopping, 数据增强)。
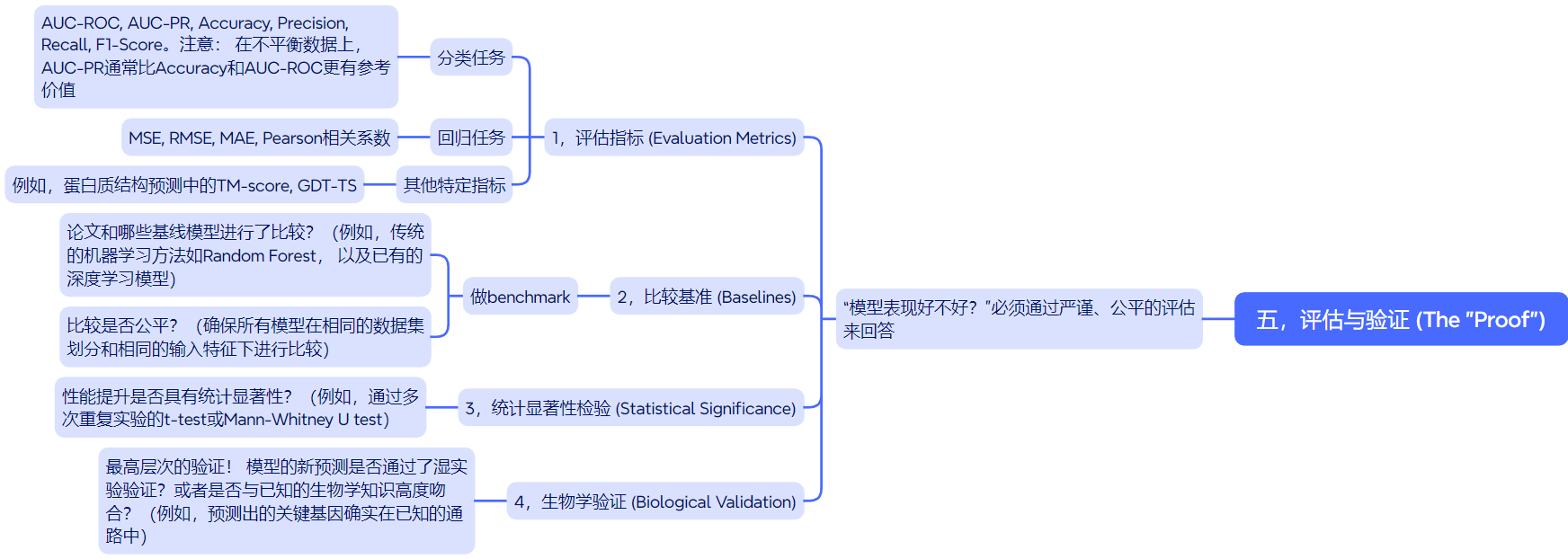
五,评估与验证 (The “Proof”)
第五层:评估与验证 (The “Proof”)
“模型表现好不好?”必须通过严谨、公平的评估来回答。
- 评估指标 (Evaluation Metrics):
- 分类任务: AUC-ROC, AUC-PR, Accuracy, Precision, Recall, F1-Score。注意: 在不平衡数据上,AUC-PR通常比Accuracy和AUC-ROC更有参考价值。
- 回归任务: MSE, RMSE, MAE, Pearson相关系数。
- 其他特定指标: 例如,蛋白质结构预测中的TM-score, GDT-TS。
- 比较基准 (Baselines): 实际上就是做benchmark
- 论文和哪些基线模型进行了比较?(例如,传统的机器学习方法如Random Forest, 以及已有的深度学习模型)。
- 比较是否公平?(确保所有模型在相同的数据集划分和相同的输入特征下进行比较)。
说白了,benchmark 就是用来评估和比较不同算法性能的重要工具,在我们这个语境下,就是比较能够用来解决某一个相同本质的生物学问题的生物学深度学习模型,一个高质量的benchmark不仅能够准确、全面地反映算法的优缺点,还能够引导整个领域的研究方向。
对于文献中展现的benchmark部分,应该注意的一些要点(仅代表本人观点):
1️⃣公平性与可重复性:也就是Fairness and reproducibility
只有在相同的条件下测试,一起比较,这样不同方法的比较才有意义:
(1)标准的数据集:所有算法要使用同1个训练集以及测试数据集
(2)统一评估流程:可能不同model在原始文献中设计的评估metrics是不一致的,我本人也遇到过需要比较一些古早文献的model,有些需要在每一轮epoch就计算一个metrics,有些是按batch,不同lab课题组开发的model,处理起来兼容性上比较麻烦,不一定好统一;
但就算是这样,我们也最好是明确各个model原本执行任务的流程和指标,尽量避免人为的干扰
(3)其他人能够复现结果,在同样的环境配置+model设计+同样的数据条件下
2️⃣评估指标要全面:
其实阅读这些文献,我们就要比较一下,文献中如果有和SOTA比较,一般比较的指标都是些什么,又是如何比较的;
又是为什么选择这个评估指标,
要知道,如果指标设计不合理,就无法准确反映算法的能力。
评估指标,要关注的是,这个指标评价的是model之间比较的什么维度,
是准确率,鲁棒性,还是效率,或者是和这个生物学问题实际需求相关的实用性(当然这个是涉及到model的可解释性的)
3️⃣多样化:
主要就是benchmark测试的场景需要切换,不能够太单一;也就是model评估比较的场景要多样化,覆盖要广。
比如说在正常序列上去评估,那么我可不可以再对突变序列、置换序列,或者是经过处理的非wild type序列进行评估;
总之就是需要契合model所设计的真实任务,其实就是变相体现model的可解释性。
- 统计显著性检验 (Statistical Significance):
- 性能提升是否具有统计显著性?(例如,通过多次重复实验的t-test或Mann-Whitney U test)。
- 生物学验证 (Biological Validation):
- 最高层次的验证! 模型的新预测是否通过了湿实验验证?或者是否与已知的生物学知识高度吻合?(例如,预测出的关键基因确实在已知的通路中)。
六,可用性与扩展性 (The “Ecosystem”)
第六层:可用性与扩展性 (The “Ecosystem”)
- 代码与模型可用性:
- 作者是否开源了代码?(GitHub)
- 是否提供了预训练模型,方便微调和部署?(Hugging Face, Model Zoo)
- 计算资源需求:
- 训练和推理需要多大的GPU内存和多长时间?这对于其他研究者的复现和使用至关重要。
- 易用性与可扩展性:
- 模型是否设计为易于在其他类似数据集上使用?是否支持迁移学习?
七,应用举例:比较两个蛋白质功能预测模型
假设你在比较模型A和模型B。
- 第一层: 它们都解决蛋白质功能预测问题(使用GO术语标签)。
- 第二层:
- A模型使用one-hot编码和PSSM特征作为输入。
- B模型使用预训练蛋白质语言模型(ESM-2)的嵌入向量作为输入。
- 它们使用相同的数据集,但B模型在划分时严格排除了与测试集有>30%序列相似性的蛋白。
- 第三层:
- A模型是一个简单的CNN。
- B模型是一个Transformer架构,并使用了在多序列比对上学到的注意力。
- 第四层: (略,假设相似)
- 第五层:
- 在测试集上,B模型的F1-Score显著高于A模型(p-value < 0.01)。
- 对B模型的注意力权重进行可视化,发现它关注了已知的功能结构域,提供了可解释性。
- 第六层:
- A和B都开源了代码,但B还提供了在线预测服务器。
通过这个框架,我们可以粗略但(比较)系统地得出结论:
B模型通过使用更先进的输入表示(预训练嵌入)、更强大的架构(Transformer)和更严谨的数据划分,实现了更高性能且具有可解释性的预测,是更优的解决方案。
这个是model解读以及计算生物学领域深度学习文献阅读的第一步。
八,model量化与比较量表:
简单将上面的这些要点进行总结,整理成一个量表其实比较方便,阅读文献就像是填表格一样(抓重点,提升效率是关键,不要lost在非主线的细枝末节上)
1. 核心元数据(模型的“名片”,用于快速索引和分类)
| 模块 | 描述 | 对比要点/问题 |
|---|---|---|
| 模型名称 | 论文中提出的模型名称 | e.g., DeepBind, AlphaFold, scBERT |
| 发表信息 | 会议/期刊、年份、代码链接 | CVPR 2023, [GitHub Link] |
| 核心问题 | 模型要解决的具体生物问题 | 转录因子结合位点预测、蛋白质结构预测、单细胞分类 |
| 任务类型 | 标准的机器学习任务分类 | 分类、回归、生成、表征学习、分割 |
2. 数据工程(Data Engineering,数据预处理)
这一块也是我们需要学习借鉴的重点部分
| 模块 | 子模块 | 对比要点/问题 |
|---|---|---|
| 输入数据 | 原始数据类型 | DNA序列、蛋白质序列、SMILES、基因表达向量、图结构、图像 |
| 数据来源 | PDB, TCGA, GEO Dataset ID: GSE12345 | |
| 特征工程 | 特征提取/编码 | One-hot, k-mer (k=?), ESM-2/ProtTrans 嵌入 (具体版本), 物理化学属性 |
| 特征维度 | 输入Tensor的最终形状 e.g., (Batch, 1000, 128) | |
| 数据预处理 | 清洗/过滤 | 去除低质量样本、长度标准化(裁剪/填充) |
| 标准化 | Z-score, Min-Max Scaling | |
| 数据集构建 | 正负样本定义 | 如何构造标签?是否存在类别不平衡? |
| 数据集划分 | 随机划分、严格的时间划分、同源排除(CD-HIT @30%)、染色体划分 | |
| 关键问题 | 是否存在数据泄露(Data Leakage)? |
3. 模型架构(Model Architecture)
技术对比的核心,可以视为一个可组装的计算图(说白了其实就是花里胡哨的什么model架构overview,考考验我们能不能够从表象中提取本质)。
其实就是架构图,可能生物学这反面,会给各种模块、各个layer起个什么乱七八糟的名字,但是我们要做的是,是直接识别出来这个是什么模板,在做什么;
比如说Fig1 overview里画了个花里胡哨的什么序列图,各种元素都标在一起,
看了method后,发现就是一个encoder,或者就是一个卷积模块(做了些特殊的处理),
那么我们这一块的目的,就是训练我们的抽象能力,直接从文献中抓取最本质的模块架构;
当然,大部分文献其实风格都比较扎实,是模块架构就是模块架构,画出来就能够直接识别。
另外,个人认为,其实model架构这一块,创新的其实是不大的,基本上用来用去就那么几个架构,然后模块缝来缝去,私以为这一块其实倒是学不到、借鉴不到什么新的东西,因为一般能够做架构创新的其实是比较少的。
| 模块 | 子模块 | 对比要点/问题 |
|---|---|---|
| 输入编码器 | 第一层网络 | 嵌入层(Embedding)、线性投影层、1D-CNN(用于捕捉局部模式) |
| 核心骨干网络 | 架构类型 | CNN, RNN (LSTM/GRU), Transformer, GNN (GAT/GCN), 混合模型 |
| 核心机制 | 使用了何种注意力?(Self-Attention, Graph Attention)、卷积核大小、层数、隐藏层维度 | |
| 输出头 | 设计 | 用于最终预测的网络层:MLP + Softmax (分类)、Linear (回归) |
| 生物先验 | 设计亮点 | 模型结构如何融入领域知识? (e.g., 用GNN模拟残基相互作用、用CNN模拟motif扫描) |
| 技术实现 | 框架 | PyTorch, TensorFlow, JAX |
| 参数量 | ~100M, ~1B |
4. 训练策略(Training Strategy)
模型的“调优手册”,决定了模型能否有效学习。
这一块,和前面的数据预处理一样,个人认为是model学习(当然是有了一定基础之后,在做工程化的积累)的另外一个关键点,比如说我们自己设计的model,能不能也仿照一下,设计一个比较新的损失函数评估等,也能够算是一个小的创新点了。
| 模块 | 子模块 | 对比要点/问题 |
|---|---|---|
| 损失函数 | 主损失 | 交叉熵损失(CE)、均方误差(MSE) |
| 自定义损失 | Focal Loss(解决不平衡)、对比损失(InfoNCE)、多任务损失 | |
| 优化器 | 选择 & 超参 | Adam (lr=1e-4), SGD with Momentum (lr=0.01, momentum=0.9) |
| 正则化 | 技术 | Dropout (rate=0.1), Weight Decay (L2=1e-5), Early Stopping, 梯度裁剪 |
| 训练技巧 | 预热 | Learning Rate Warmup |
| 调度器 | Cosine Annealing, ReduceLROnPlateau | |
| 预训练 | 策略 | 是否进行预训练?掩码语言模型(MLM)、自监督对比学习 |
当然,还有一点需要注意的就是,调超参毕竟也是门学问,这么多年了,其实有很多经验以及工程化的技术都已经相对比较成熟了,比如说使用optuna来进行超参数的优化,简单俩说就是加入调节超参数的超参数,
比如说设置XGBoost或者是lightGBM的learning rate多少到多少、然后树也就是弱学习器要多少个,或者是树的深度有深,然后叶节点个数等,或者是L1以及L2正则化参数设置的范围多少到多少等等。
其实这些也可以记录下来。
5. 评估与验证(Evaluation & Validation)
用于公平地衡量模型的“性能”和“价值”,要点一般就按照前面提到的benchmark的要求来;
当然,这里其实也涉及到一个问题,就是到哪里寻找我们所研究问题的SOTA模型,如果是解决比较general的问题,比如说是蛋白质结构的folding问题(序列到结构)、inverse folding(结构到序列)、domain segmentation(结构域划分,本质是注释问题)等等,如果是general的问题,一般是能够找到专门收集这些domain内的SOTA模型的数据库,会有专人收集。
如果是比较小众的问题,
很多情况下,可能就需要阅读文献了,当然实际情况会复杂一点:
比如说个人曾经遇到过这么一种情况:
解决某一个特定的小问题,比如说是某种特殊的基因编辑之后的序列比对,
需要开发一些特殊的工具,
这个时候有些文献可能也是专门做这个的,那好,好在我们还不需要做比较heavy的工程化任务;
但是有些文献可能是开发了一个大套件、大软件,然后其中部分流程处理是分配到这个问题上的,这个时候有代码、开源的还好,我们只需要读懂别人的源码以及软件设计的逻辑,然后将这一部分业务代码剥离出来,方便统一做benchmark;
如果没有open source,那么可能就需要我们自己在阅读理解文献之后,凭借过硬的数学逻辑能力、与过硬的计算机工程化能力,为别人构建一个假想的轮子了(这种情况是真有可能发生的)
| 模块 | 子模块 | 对比要点/问题 |
|---|---|---|
| 评估指标 | 主要指标 | AUC-ROC, AUC-PR, Accuracy, F1-Score, MSE, MAE |
| 领域指标 | TM-score, RMSD (结构预测) | |
| 基线对比 | 对比模型 | 和哪些SOTA或基线模型比较?(e.g., Random Forest, previous DL model) |
| 公平性 | 是否在相同的数据划分和相同的输入特征下比较? | |
| 显著性检验 | 方法 | 是否报告了多次运行的均值和标准差?是否进行了t-test? |
| 可解释性 | 方法 | 注意力权重可视化、显著图(Saliency Map)、特征重要性,比如说是烂大街的SHAP |
| 生物学验证 | 模型发现的特征是否对应已知生物学知识?(e.g., 识别出已知的蛋白 motif) |
6. 部署与可用性(Ops & Usability)
| 模块 | 子模块 | 对比要点/问题 |
|---|---|---|
| 代码开源 | 可用性 | 代码是否公开?(是/否/部分) |
| 代码质量 | 文档是否完善?是否易于复现? | |
| 模型可用性 | 预训练权重 | 是否提供?方便下游任务微调(Fine-tune) |
| 推理脚本 | 是否提供完整的预测示例? | |
| 计算成本 | 训练资源 | 需要多少GPU小时?何种型号GPU?(e.g., 8x A100, 7 days) |
| 推理速度 | 每秒能处理多少样本?能否满足实时需求? | |
| 可扩展性 | 设计 | 模型是否模块化?是否易于移植到新数据或类似任务? |
九,量表模板
仅作为参考,重点是有针对性的阅读与model比较,形式可以千变万化。
主题: [在此填写你的研究主题,例如:蛋白质结构预测模型比较]
1. 模型元信息 (Meta Information)
| 模型名称 | 发表年份 | 会议/期刊 | 代码链接 | 核心问题 | 任务类型 |
|---|---|---|---|---|---|
Model A | 2021 | Nature | [GitHub Link] | 蛋白质功能预测 | 多标签分类 |
Model B | 2022 | ICML | [GitHub Link] | 蛋白质功能预测 | 多标签分类 |
[你的模型] | - | - | - | - | - |
| … |
2. 数据与特征 (Data & Features)
| 模型名称 | 输入数据 | 特征编码 | 数据集划分策略 | 数据来源/ID | 关键问题 (e.g., 数据泄露?) |
|---|---|---|---|---|---|
Model A | 蛋白质序列 | One-hot + PSSM | 随机划分 | UniProt | 可能存在同源泄露 |
Model B | 蛋白质序列 | ESM-2 嵌入 (650M) | 严格同源排除 (30%) | UniProt | 划分严谨 |
[你的模型] | |||||
| … |
3. 模型架构 (Model Architecture)
| 模型名称 | 核心骨干网络 | 核心机制/注意力 | 生物先验设计 | 参数量 | 实现框架 |
|---|---|---|---|---|---|
Model A | 1D-CNN | N/A | CNN捕捉局部motif | ~1M | TensorFlow |
Model B | Transformer | Self-Attention | 注意力权重可解释 | ~650M | PyTorch |
[你的模型] | |||||
| … |
4. 训练策略 (Training Strategy)
| 模型名称 | 损失函数 (Loss) | 优化器 & 学习率 | 正则化技术 | 预训练策略 | 训练硬件/时间 |
|---|---|---|---|---|---|
Model A | Binary Cross-Entropy | Adam (lr=1e-3) | Dropout (0.5) | 无 | 1x V100, 4h |
Model B | Focal Loss | AdamW (lr=5e-5) | Weight Decay | MLM on UniRef50 | 8x A100, 1周 |
[你的模型] | |||||
| … |
5. 性能与评估 (Performance & Evaluation)
| 模型名称 | 主要指标 (Main Metric) | 基线对比 (vs. SOTA) | 统计显著性 | 可解释性分析 | 生物学验证 |
|---|---|---|---|---|---|
Model A | AUC-PR: 0.75 | 比 LR 提升 +5% | 未报告 | Saliency Maps | 与已知 motif 部分吻合 |
Model B | AUC-PR: 0.92 | 比 Model A 提升 +17% | p < 0.01 | Attention Visualization | 预测结果被实验验证 |
[你的模型] | |||||
| … |
6. 可用性与总结 (Usability & Conclusion)
| 模型名称 | 代码可用性 | 模型可用性 (预训练权重) | 计算成本 (推理) | 核心优势 | 主要局限 |
|---|---|---|---|---|---|
Model A | 代码开源 | 无 | 低,速度快 | 模型简单,易部署 | 性能一般,泛化能力弱 |
Model B | 代码开源 | 提供 checkpoint | 高,需要GPU | SOTA性能,可解释性强 | 计算资源要求高 |
[你的模型] | |||||
| … |