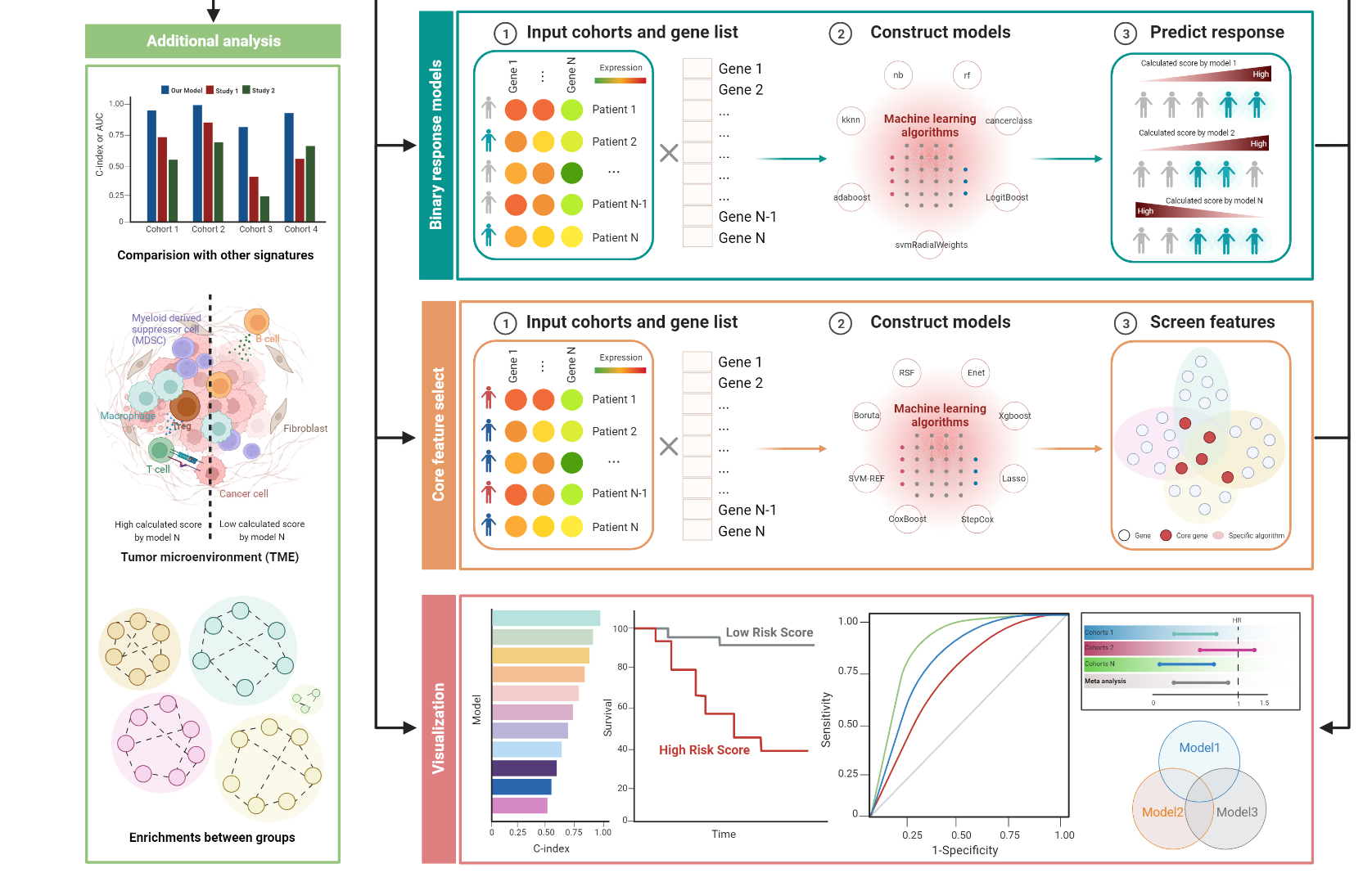
基于R语言,“上百种机器学习模型”学习教程 | Mime包
原文链接:基于R语言,“上百种机器学习模型”学习教程 | Mime包【点击访问】
本期教程
Citations
- Liu H, Zhang W, Zhang Y, Adegboro AA, Fasoranti DO, Dai L, Pan Z, Liu H, Xiong Y, Li W, Peng K, Wanggou S, Li X. Mime: A flexible machine-learning framework to construct and visualize models for clinical characteristics prediction and feature selection. Comput Struct Biotechnol J. 2024.

# 链接:
https://pmc.ncbi.nlm.nih.gov/articles/PMC11269309/
- Liu H, Zhang W, Zhang, Y, Li X, Wanggou S. Constructing and Visualizing Models using Mime-based Machine-learning Framework. J Vis Exp. 2025.

# 链接:
https://app.jove.com/t/68553/constructing-visualizing-models-using-mime-based-machine-learning
安装
# options("repos"= c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
# options(BioC_mirror="http://mirrors.tuna.tsinghua.edu.cn/bioconductor/")
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")depens<-c('GSEABase', 'GSVA', 'cancerclass', 'mixOmics', 'sparrow', 'sva' , 'ComplexHeatmap' )
for(i in 1:length(depens)){depen<-depens[i]if (!requireNamespace(depen, quietly = TRUE)) BiocManager::install(depen,update = FALSE)
}if (!requireNamespace("CoxBoost", quietly = TRUE))devtools::install_github("binderh/CoxBoost")if (!requireNamespace("fastAdaboost", quietly = TRUE))devtools::install_github("souravc83/fastAdaboost")if (!requireNamespace("Mime", quietly = TRUE))devtools::install_github("l-magnificence/Mime")library(Mime1)
Mime1很难安装,个人测试。
Used
1. 文件需要
Mime需要多个队列,其中包含转录测序数据,以及生存或临床对治疗反应的信息,以及作为输入的基因集。您可以在 External Data File 中下载示例数据,以检查输入的格式。
load("./Example.cohort.Rdata")
list_train_vali_Data[["Dataset1"]][1:5,1:5]
#> ID OS.time OS MT-CO1 MT-CO3
#> TCGA.DH.A66B.01 1281.65322 0 13.77340 13.67931
#> TCGA.HT.7607.01 96.19915 1 14.96535 14.31857
#> TCGA.DB.A64Q.01 182.37755 0 13.90659 13.65321
#> TCGA.DU.8167.01 471.97707 0 14.90695 14.59776
#> TCGA.HT.7610.01 1709.53901 0 15.22784 14.62756
第一列是样本 ID, 第二至第三列OS.time和OS是患者的生存时间和状态,其他列是用log2(x+1)标度的基因表达水平。Dataset1是训练数据集,其他 Dataset 是验证数据集。
load("./Example.ici.Rdata")
list_train_vali_Data[["training"]][1:5,1:5]
#> ID Var FTH1 EEF1A1 ACTB
#> SAMf2ce197162ce N 10.114846 4.817746 11.230180
#> ERR2208915 Y 2.044180 5.038854 3.977902
#> G138701_RCCBMS-00141-T_v1_RNA_OnPrem Y 5.406008 5.341635 5.366668
#> SAMe41b1e773582 N 9.215794 4.707360 11.412721
#> SAM5ffd7e4cd794 N 9.003710 3.908884 10.440559
第一列是样本ID, 第二列Var是患者的治疗反应 (N:无反应;Y:反应), 其他列是log2(x+1)标度的基因表达水平。训练是训练数据集,其他数据集是验证。
load("./genelist.Rdata")
#> [1] "MYC" "CTNNB1" "JAG2" "NOTCH1" "DLL1" "AXIN2" "PSEN2" "FZD1" "NOTCH4" "LEF1" "AXIN1" "NKD1" "WNT5B"
#>[14] "CUL1" "JAG1" "MAML1" "KAT2A" "GNAI1" "WNT6" "PTCH1" "NCOR2" "DKK4" "HDAC2" "DKK1" "TCF7" "WNT1"
#>[27] "NUMB" "ADAM17" "DVL2" "PPARD" "NCSTN" "HDAC5" "CCND2" "FRAT1" "CSNK1E" "RBPJ" "FZD8" "TP53" "SKP2"
#>[40] "HEY2" "HEY1" "HDAC11"
这个基因组与MSigDB的Wnt/β-catenin信号传导有关。
We recommend training dataset with more than 100 samples and gene set with more than 50 genes.
1. 构建预后预测模型
选择最优模型
library(Mime1)
load("./Example.cohort.Rdata")
load("./genelist.Rdata")
res <- ML.Dev.Prog.Sig(train_data = list_train_vali_Data$Dataset1,list_train_vali_Data = list_train_vali_Data,unicox.filter.for.candi = T,unicox_p_cutoff = 0.05,candidate_genes = genelist,mode = 'all',nodesize =5,seed = 5201314 )
ML.Dev.Prog.Sig()提供了三种模式,包括all、single和double。ALL:标识使用所有十种算法及其组合。Single:表示只使用十种算法中的一种。double:表示使用两种算法的组合。在大多数情况下,我们通常会使用all模式来分析数据。- 如果你将
unicox.filter.for.candi设置为T (默认值),Mime首先会在训练数据集中对提供的基因进行单变量cox回归,以筛选出预后变量,然后用于构建模型。
绘制每个模型的C-index:
cindex_dis_all(res,validate_set = names(list_train_vali_Data)[-1],
order =names(list_train_vali_Data),width = 0.35)
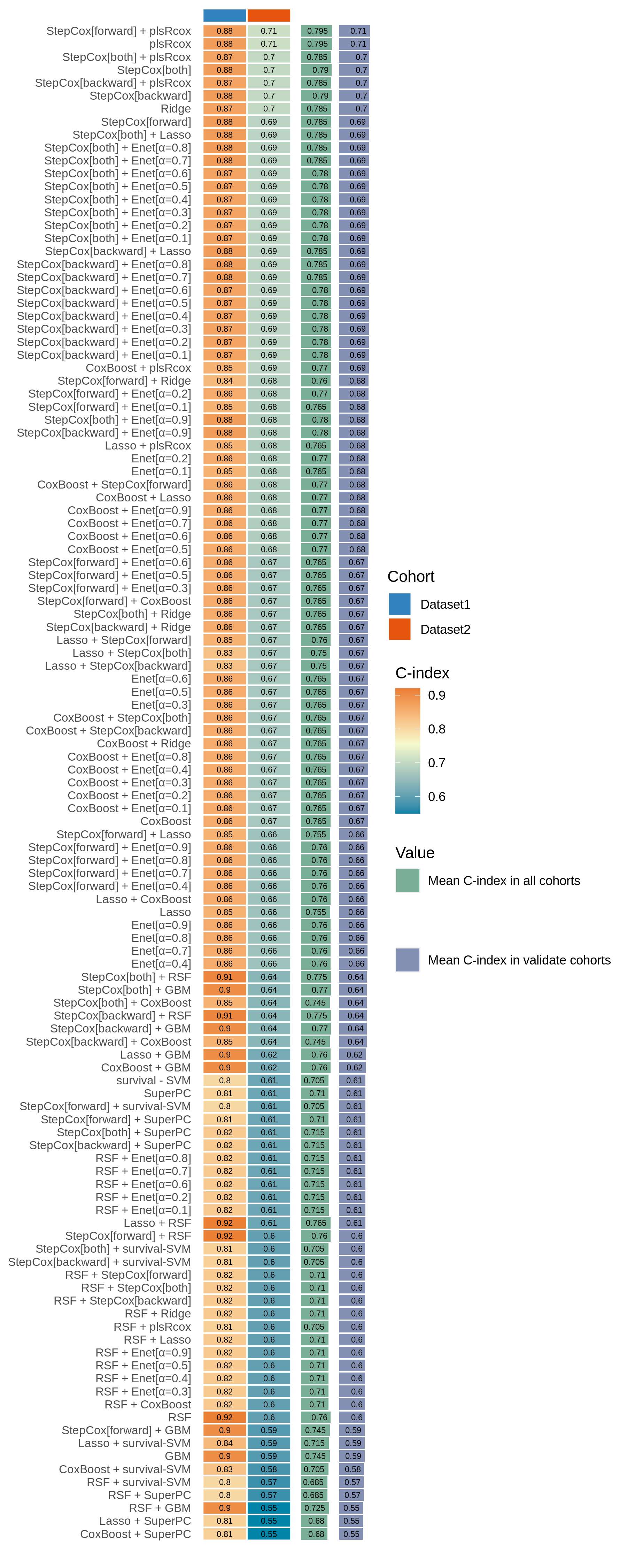
在不同数据集之间绘制特定模型的C-index
cindex_dis_select(res,model="StepCox[forward] + plsRcox",order= names(list_train_vali_Data))
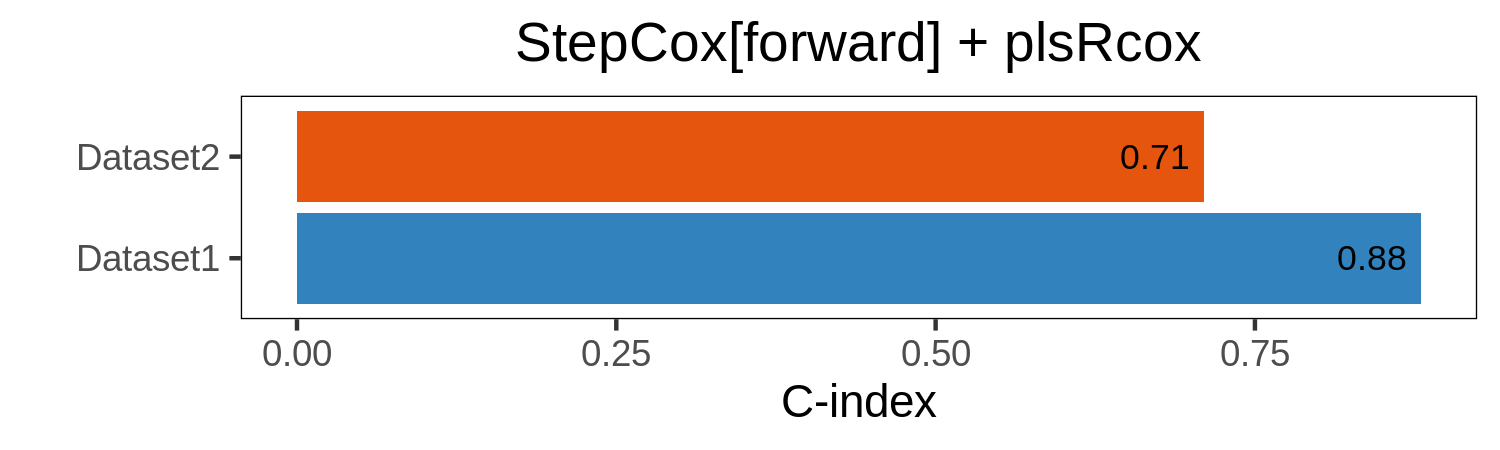
如果输入对象res来自ML.Dev.Prog.Sig()中使用的所有模型,则应将model定义为特定的模型名称,同时将model定义为SOD。
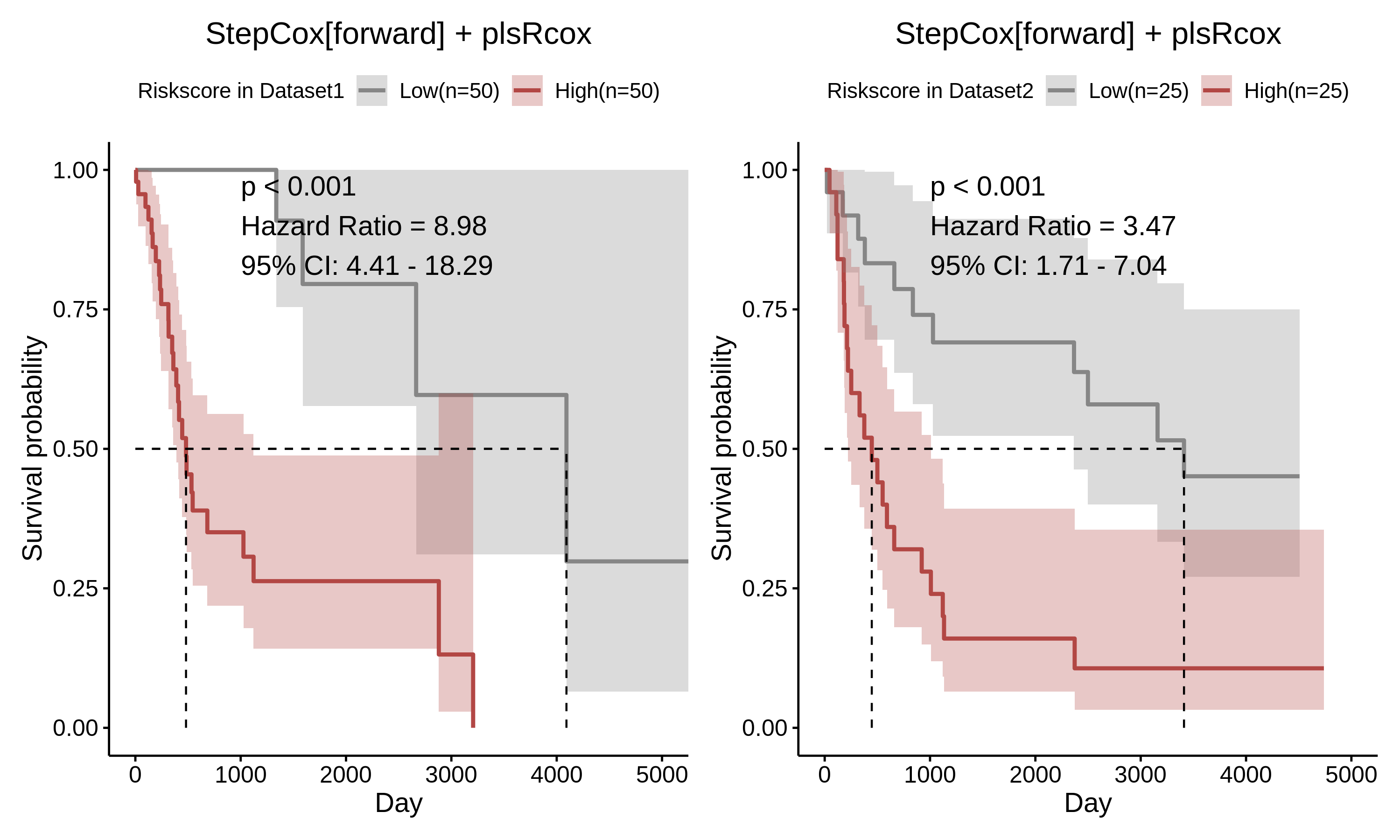
根据特定模型在不同数据集中计算的风险评分绘制患者生存曲线:
survplot <- vector("list",2)
for (i in c(1:2)) {print(survplot[[i]]<-rs_sur(res, model_name = "StepCox[forward] + plsRcox",dataset = names(list_train_vali_Data)[i],#color=c("blue","green"),median.line = "hv",cutoff = 0.5,conf.int = T,xlab="Day",pval.coord=c(1000,0.9)))
}
aplot::plot_list(gglist=survplot,ncol=2)
1.2 计算每个模型的AUC scores
all.auc.1y <- cal_AUC_ml_res(res.by.ML.Dev.Prog.Sig = res,train_data = list_train_vali_Data[["Dataset1"]],inputmatrix.list = list_train_vali_Data,mode = 'all',AUC_time = 1,auc_cal_method="KM")
all.auc.3y <- cal_AUC_ml_res(res.by.ML.Dev.Prog.Sig = res,train_data = list_train_vali_Data[["Dataset1"]],inputmatrix.list = list_train_vali_Data,mode = 'all',AUC_time = 3,auc_cal_method="KM")
all.auc.5y <- cal_AUC_ml_res(res.by.ML.Dev.Prog.Sig = res,train_data = list_train_vali_Data[["Dataset1"]],inputmatrix.list = list_train_vali_Data,mode = 'all',AUC_time = 5,auc_cal_method="KM")
Cal_AUC_ml_res()提供了三种模式,这些模式应该与 ML.Dev.Prog.Sig () 使用的模式一致。AUC_timefor 1 year, 2 years, 3 years…, We recommend using the shortest survival time among all datasets.
only plot 1-year AUC predicted by all models:
auc_dis_all(all.auc.1y,dataset = names(list_train_vali_Data),validate_set=names(list_train_vali_Data)[-1],order= names(list_train_vali_Data),width = 0.35,year=1)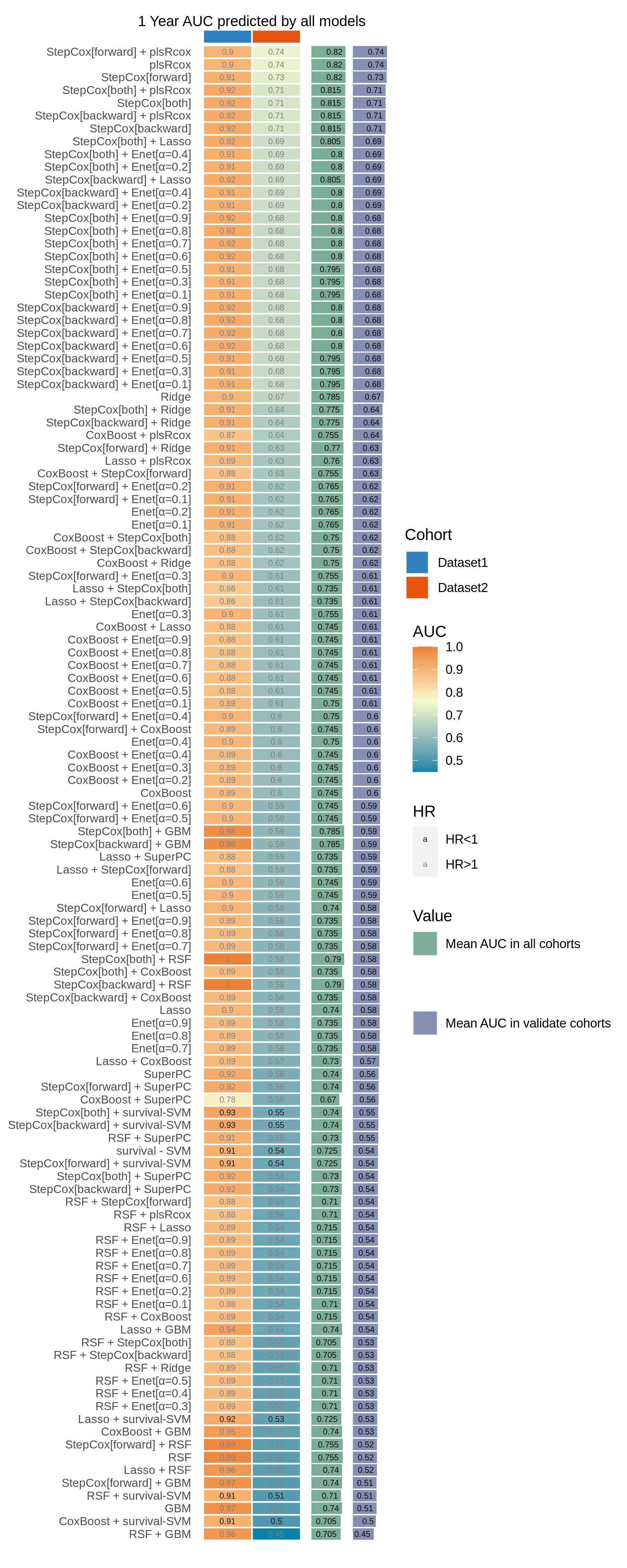
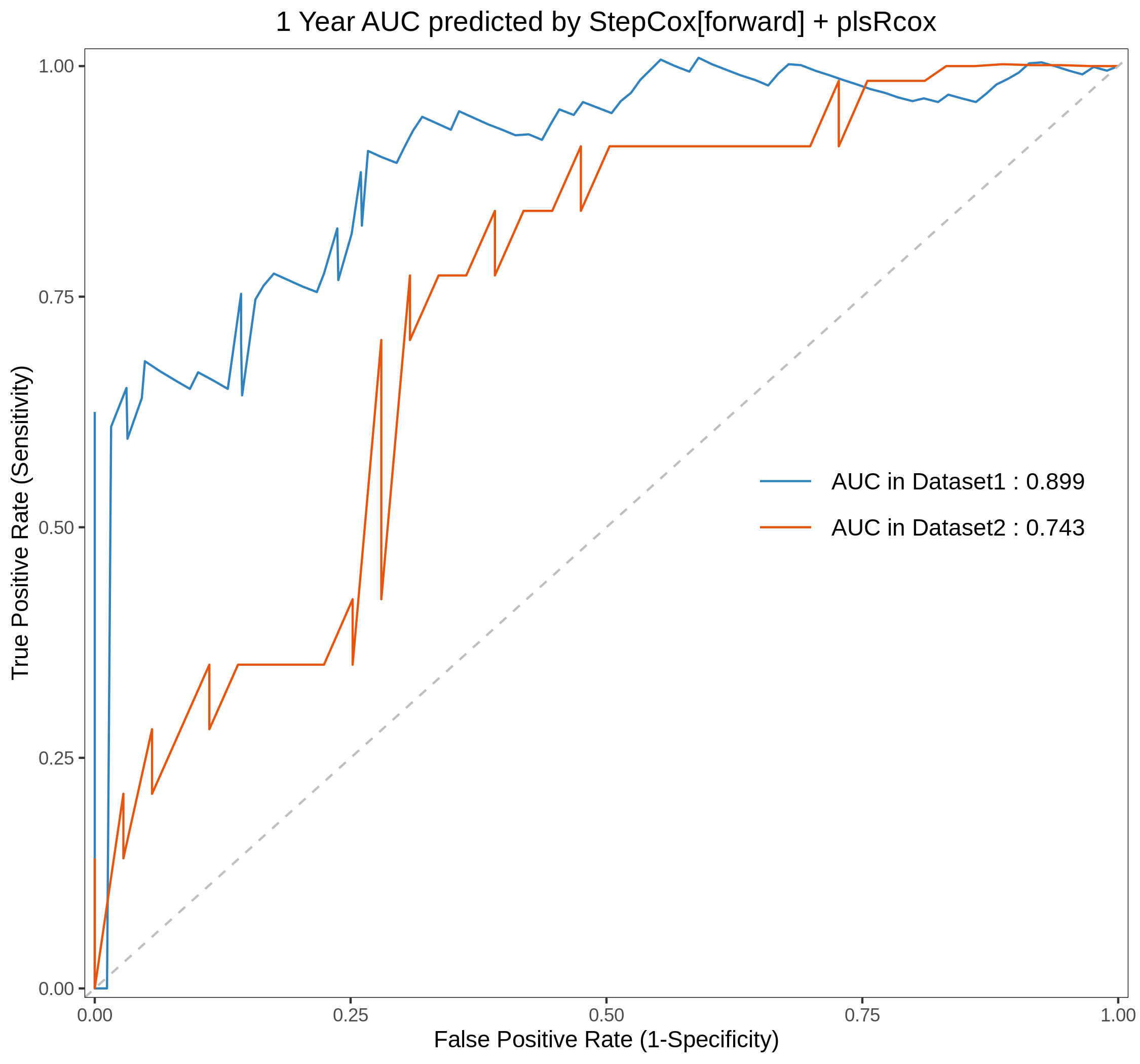
绘制不同数据集中特定模型的ROC图
roc_vis(all.auc.1y,model_name = "StepCox[forward] + plsRcox",dataset = names(list_train_vali_Data),order= names(list_train_vali_Data),anno_position=c(0.65,0.55),year=1)
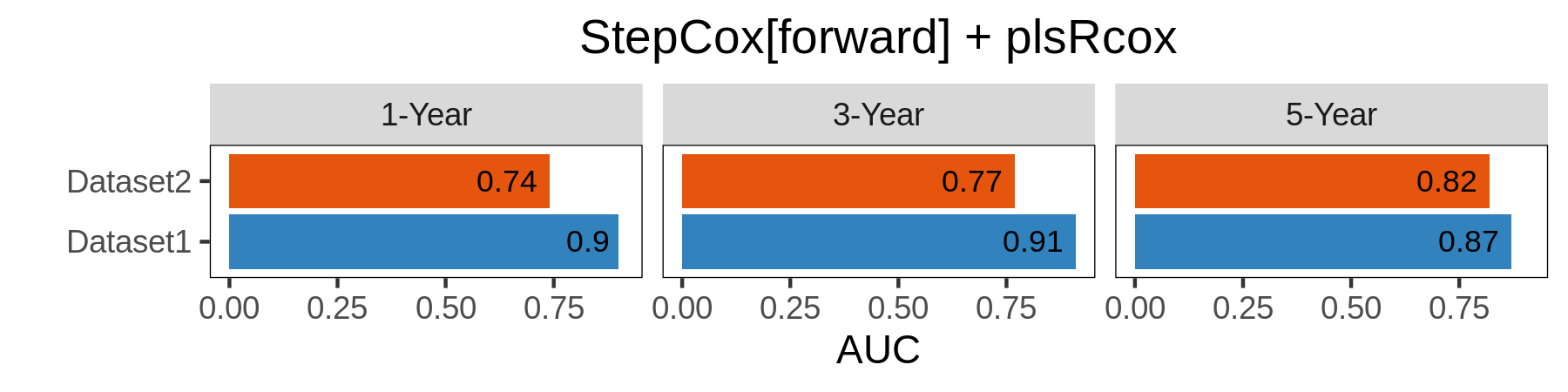
Plot 1, 3, and 5-year AUC of specific model among different datasets:
auc_dis_select(list(all.auc.1y,all.auc.3y,all.auc.5y),model_name="StepCox[forward] + plsRcox",dataset = names(list_train_vali_Data),order= names(list_train_vali_Data),year=c(1,3,5))
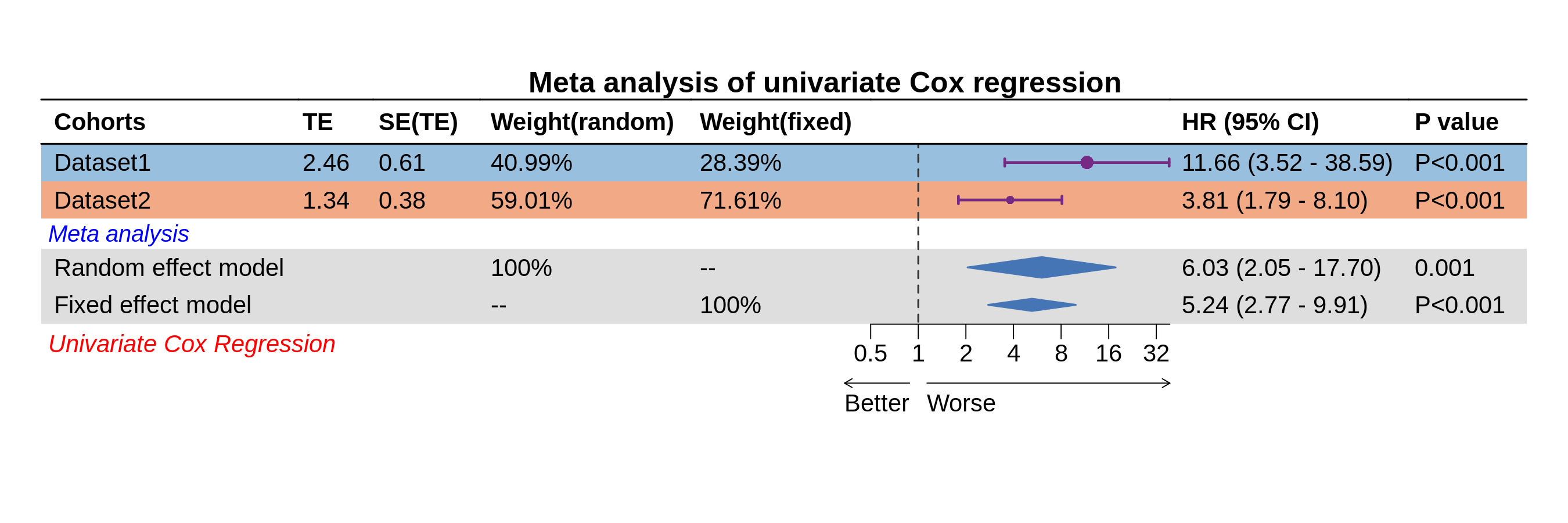
1.3 特定模型的单变量 Cox 回归元分析
unicox.rs.res <- cal_unicox_ml_res(res.by.ML.Dev.Prog.Sig = res,optimal.model = "StepCox[forward] + plsRcox",type ='categorical')
metamodel <- cal_unicox_meta_ml_res(input = unicox.rs.res)
meta_unicox_vis(metamodel,dataset = names(list_train_vali_Data))
typeincludescategoricalandcontinuous.
1.4 与先前发表的模型进行比较
rs.glioma.lgg.gbm <- cal_RS_pre.prog.sig(use_your_own_collected_sig = F,type.sig = c('LGG','GBM','Glioma'),list_input_data = list_train_vali_Data)
cal_RS_pre.prog.sig()will calculate the risk score based on the signatures from previous papers.
特定模型的HR与之前发表的模型进行比较:
HR_com(rs.glioma.lgg.gbm,res,model_name="StepCox[forward] + plsRcox",dataset=names(list_train_vali_Data),type = "categorical")
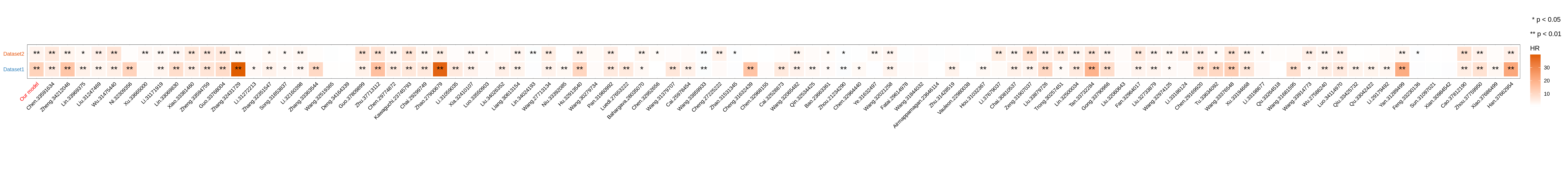
cc.glioma.lgg.gbm <- cal_cindex_pre.prog.sig(use_your_own_collected_sig = F,type.sig = c('Glioma','LGG','GBM'),list_input_data = list_train_vali_Data)
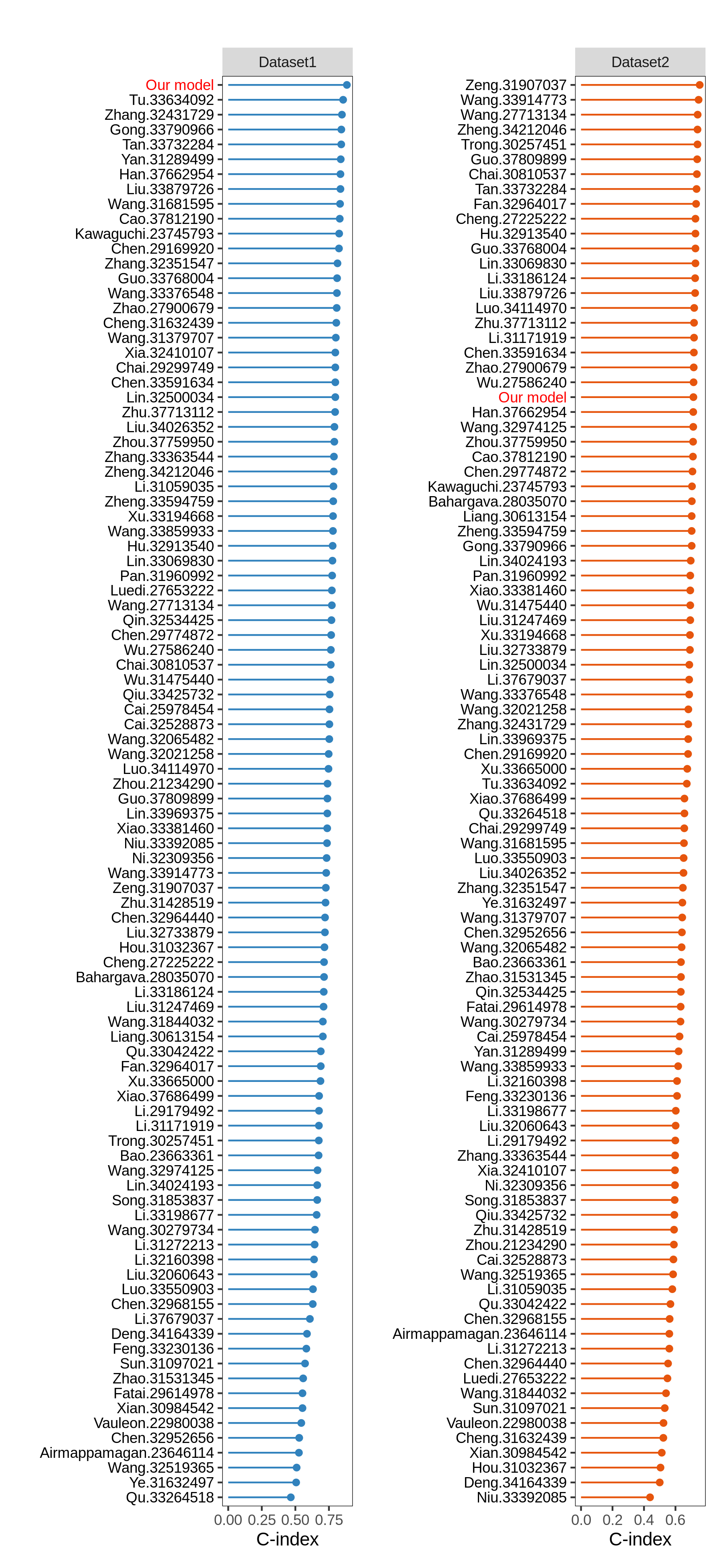
cal_cindex_pre.prog.sig()will calculate the C-index based on the signatures from previous papers like the fuctioncal_RS_pre.prog.sig().
将特定模型的C-index与之前发表的模型进行比较:
cindex_comp(cc.glioma.lgg.gbm,res,model_name="StepCox[forward] + plsRcox",dataset=names(list_train_vali_Data))
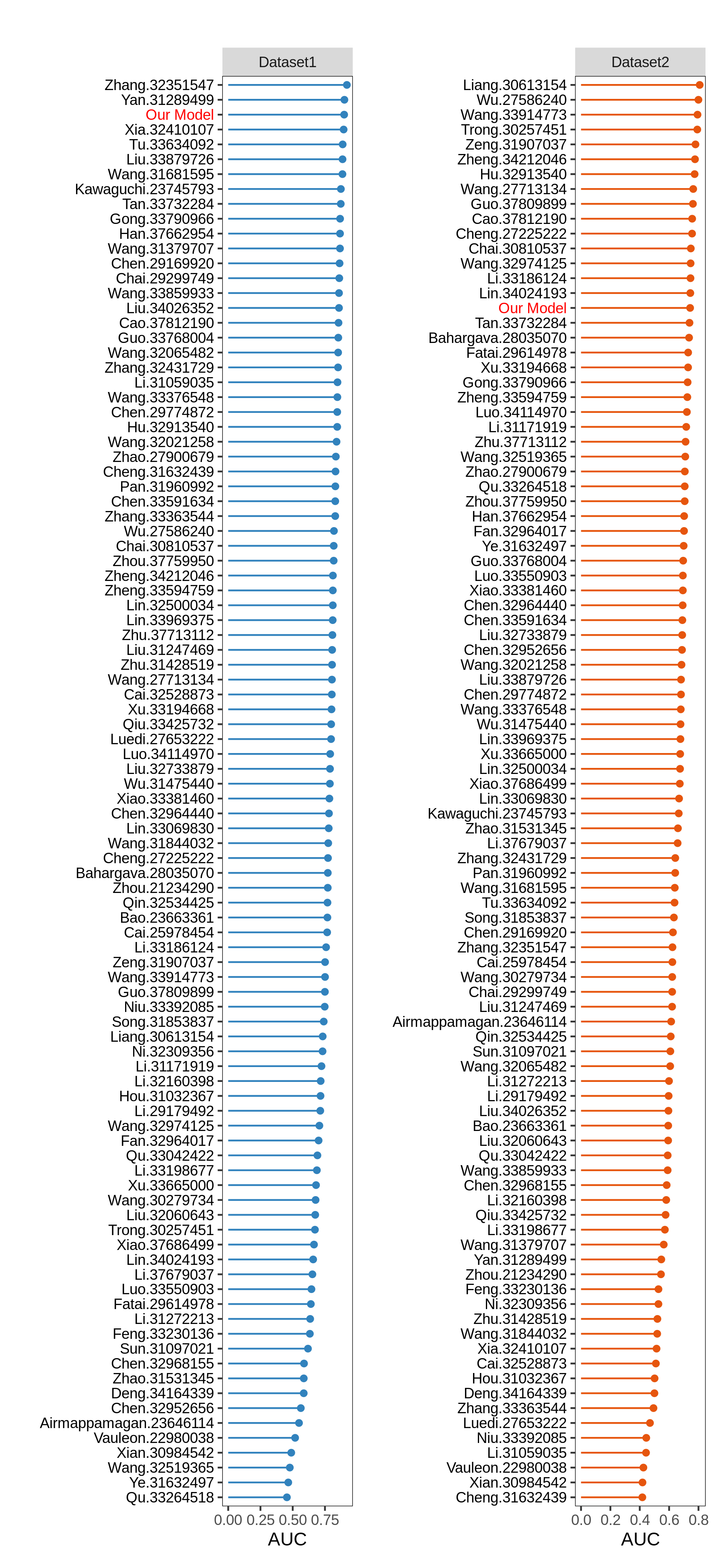
auc.glioma.lgg.gbm.1 <- cal_auc_pre.prog.sig(use_your_own_collected_sig = F,type.sig = c('Glioma','LGG','GBM'),list_input_data = list_train_vali_Data,AUC_time = 1,auc_cal_method = 'KM')
Call_auc_pre.prog.sig()将基于以前论文的签名计算 AUC, 例如cal_RS_pre.prog.sig()函数。AUC_time类似于cal_AUC_ml_res()的要求。
与特定模型的AUC与之前发布的模型进行比较:
auc_comp(auc.glioma.lgg.gbm.1,all.auc.1y,model_name="StepCox[forward] + plsRcox",dataset=names(list_train_vali_Data))
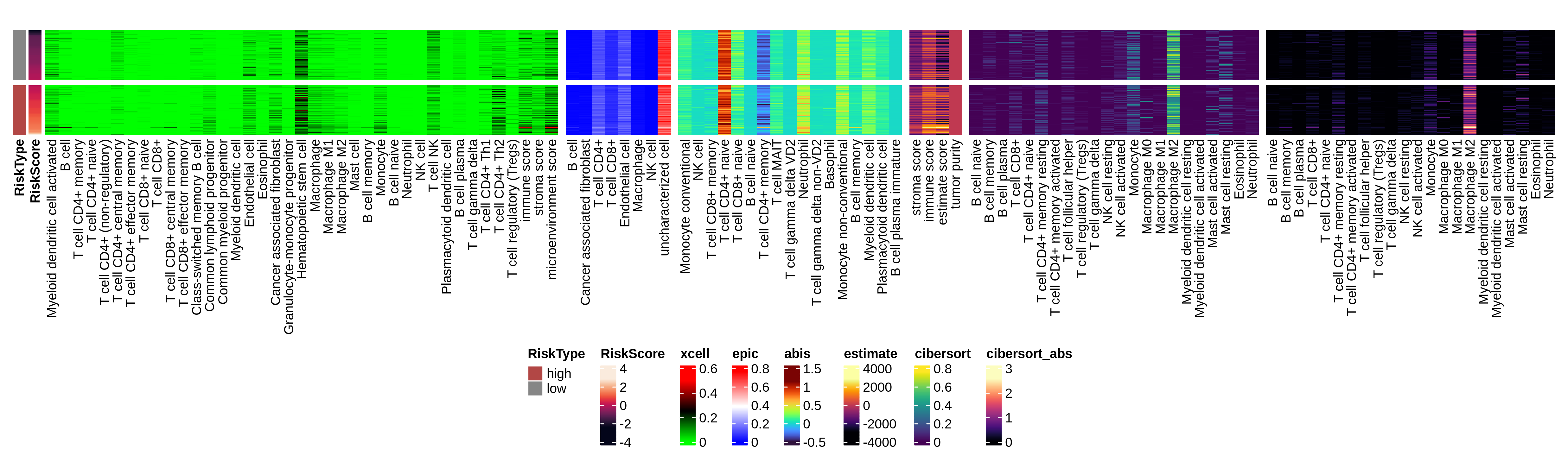
1.5 免疫浸润分析
devo <- TME_deconvolution_all(list_train_vali_Data)
- TME_deconvolution_all() includes 10 deconvolution methods (“quantiseq”, “xcell”, “epic”, “abis”, “mcp_counter”, “estimate”, “cibersort”, “cibersort_abs”, “timer”, “consensus_tme”) from immunedeconv::deconvolution_methods. By default, deconvolution methods are set as (“xcell”, “epic”, “abis”, “estimate”, “cibersort”, “cibersort_abs”).
immuno_heatmap(res,devo,model_name="StepCox[backward] + plsRcox",dataset="Dataset1")
2. 构建响应的预测模型
load("./Example.ici.Rdata")
load("./genelist.Rdata")
res.ici <- ML.Dev.Pred.Category.Sig(train_data = list_train_vali_Data$training,list_train_vali_Data = list_train_vali_Data,candidate_genes = genelist,methods = c('nb','svmRadialWeights','rf','kknn','adaboost','LogitBoost','cancerclass'),seed = 5201314,cores_for_parallel = 60
)
ML.Dev.Pred.Category.Sig()使用机器学习算法开发二进制变量的预测模型。
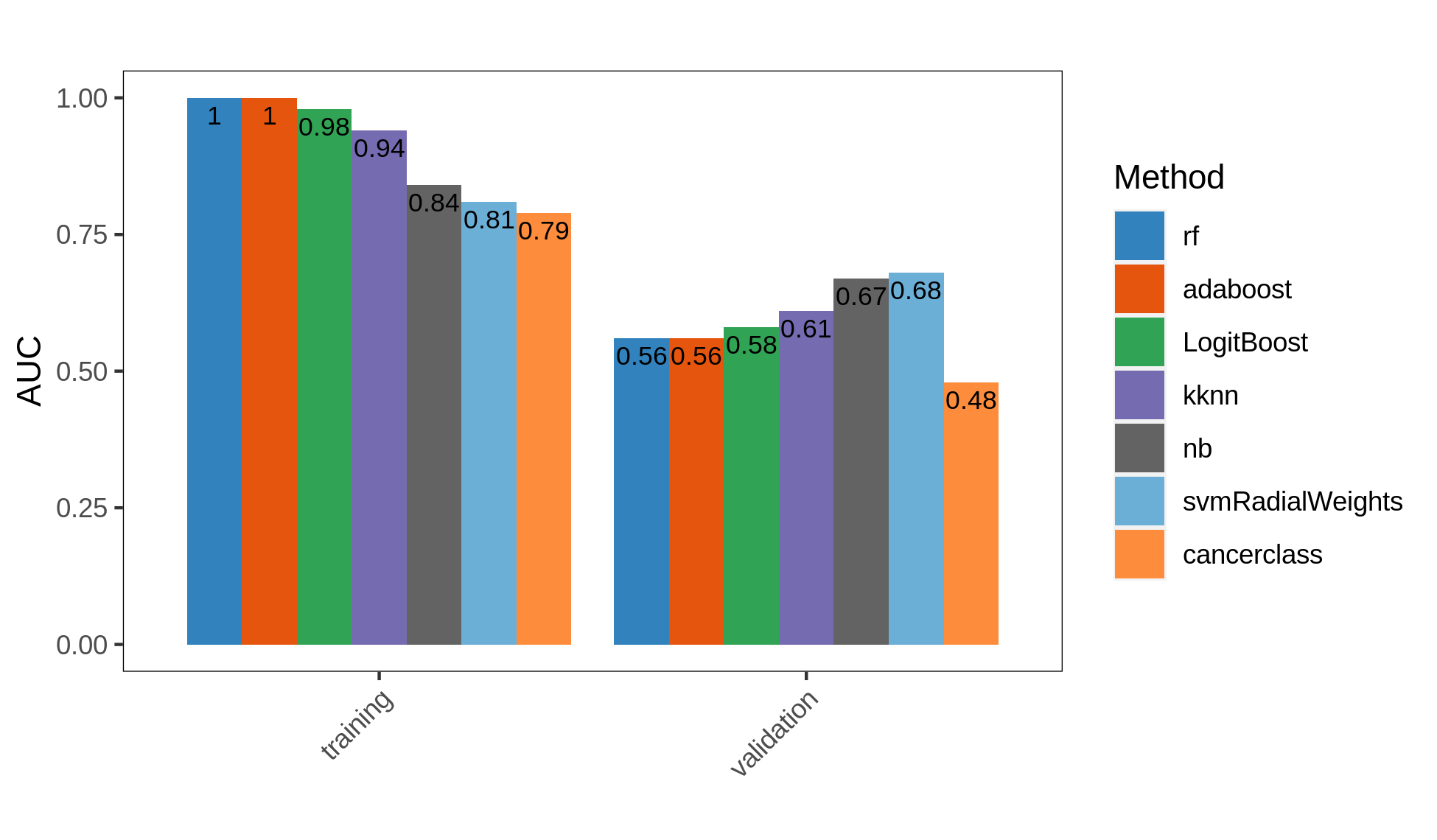
绘制不同数据集中不同方法的AUC:
auc_vis_category_all(res.ici,dataset = c("training","validation"),order= c("training","validation"))
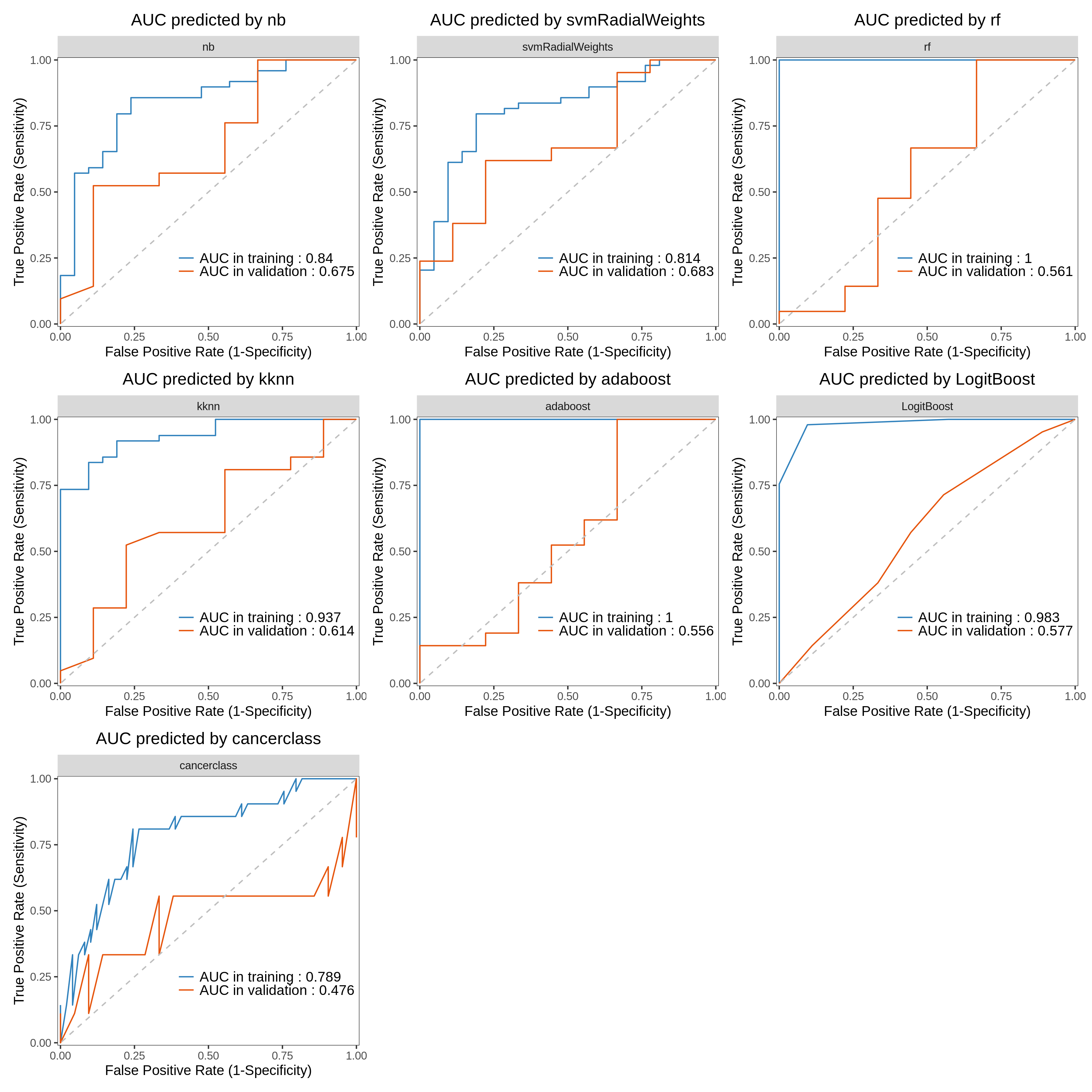
绘制特定方法在不同数据集之间的ROC:
plot_list<-list()
methods <- c('nb','svmRadialWeights','rf','kknn','adaboost','LogitBoost','cancerclass')
for (i in methods) {plot_list[[i]]<-roc_vis_category(res.ici,model_name = i,dataset = c("training","validation"),order= c("training","validation"),anno_position=c(0.4,0.25))
}
aplot::plot_list(gglist=plot_list,ncol=3)
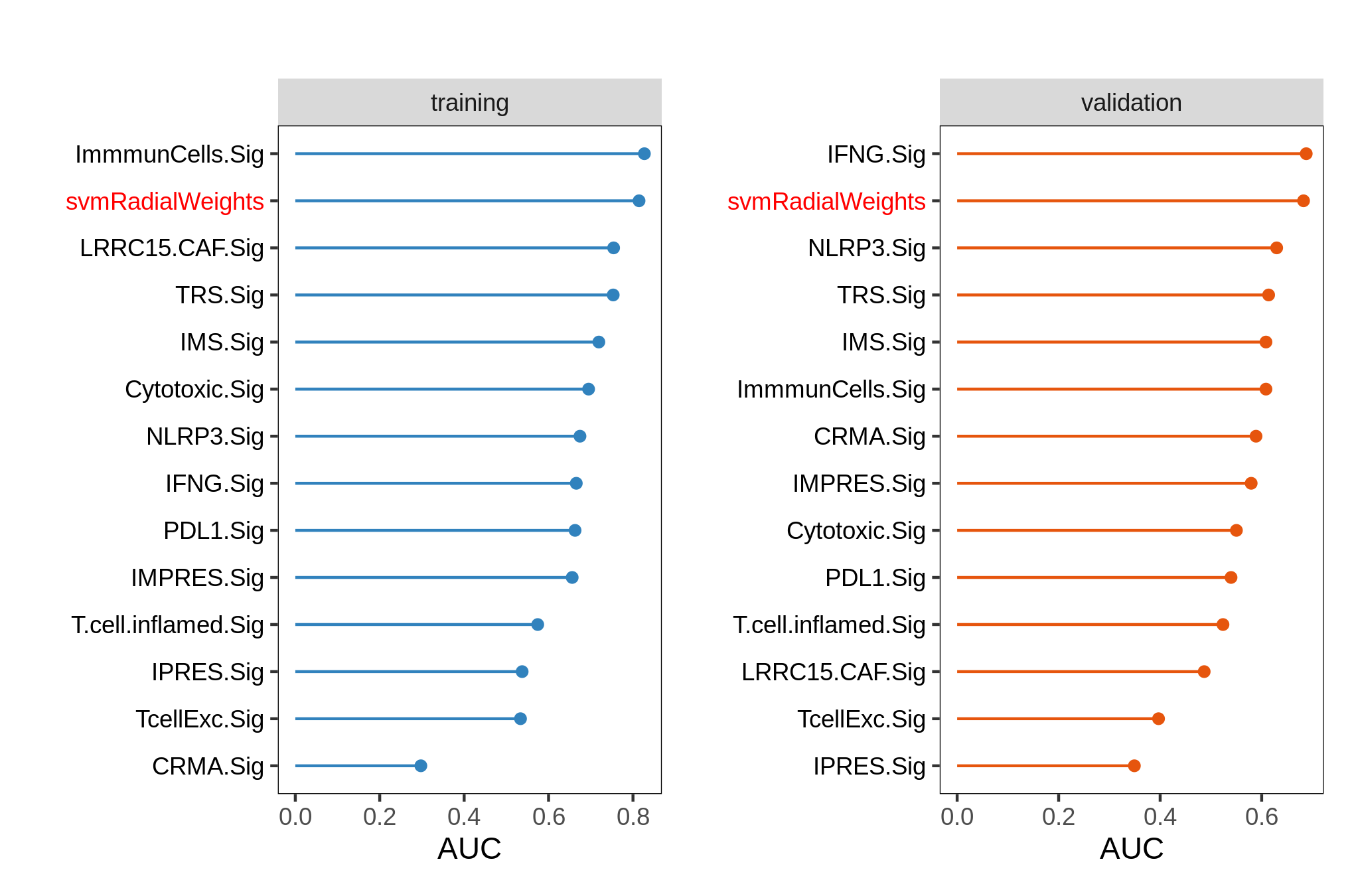
将AUC与其他已发表的与免疫治疗反应相关的模型进行比较:
auc.other.pre <- cal_auc_previous_sig(list_train_vali_Data = list_train_vali_Data,seed = 5201314,train_data = list_train_vali_Data$training,cores_for_parallel = 32)
cal_auc_previous_sig(): 将根据先前论文的签名计算免疫治疗反应的AUC。cores_for_parallel是指您可以选择用于并行操作的核心。如果多核心条件是错误的,请将cores_for_parallel设置为 1。
绘制特定模型的比较结果图:
auc_category_comp(res.ici,auc.other.pre,model_name="svmRadialWeights",dataset=names(list_train_vali_Data))
3. 核心特征选择
load("./Example.cohort.Rdata")
load("./genelist.Rdata")
res.feature.all <- ML.Corefeature.Prog.Screen(InputMatrix = list_train_vali_Data$Dataset1,candidate_genes = genelist,mode = "all",nodesize =5,seed = 5201314 )
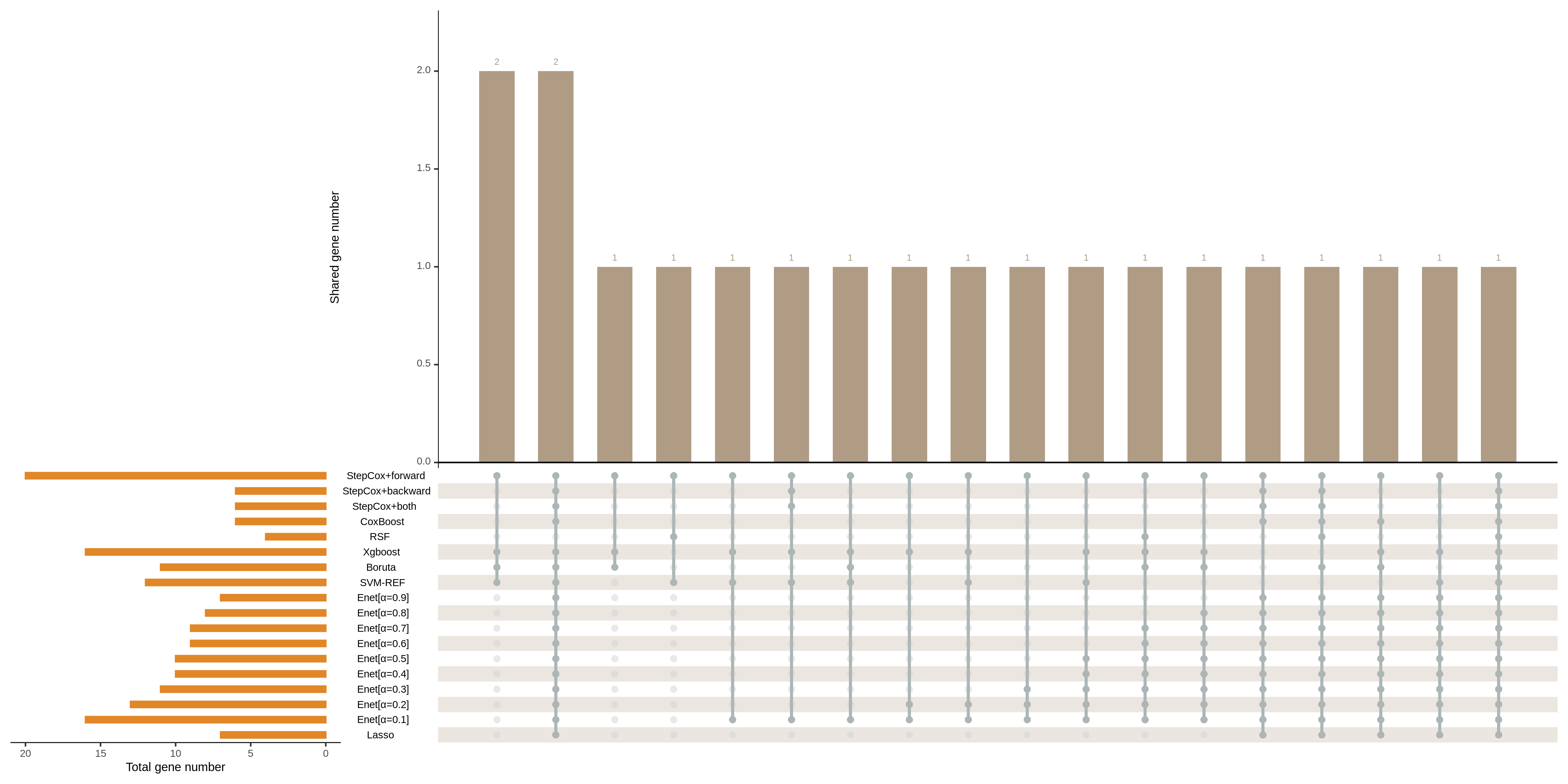
ML.Corefeature.Prog.Screen()提供了三种模式,包括all、single和all_without_SVM。all模式意味着使用所有八种方法 (“RSF”、“Enet”、“Boruta”、“Xgboost”、“SVM-REF”、“Lasso”、“CoxBoost”、“StepCox”) 进行选择。single模式意味着只使用一种方法进行运行。如果使用single模式,则应在八种方法中指定single_ml。由于SVM耗时太长,我们定义了其他七种用于选择的方法作为all_without_SVM模式。
通过不同方法筛选的基因的Upset plot:
core_feature_select(res.feature.all)
绘制不同方法过滤的基因序列:
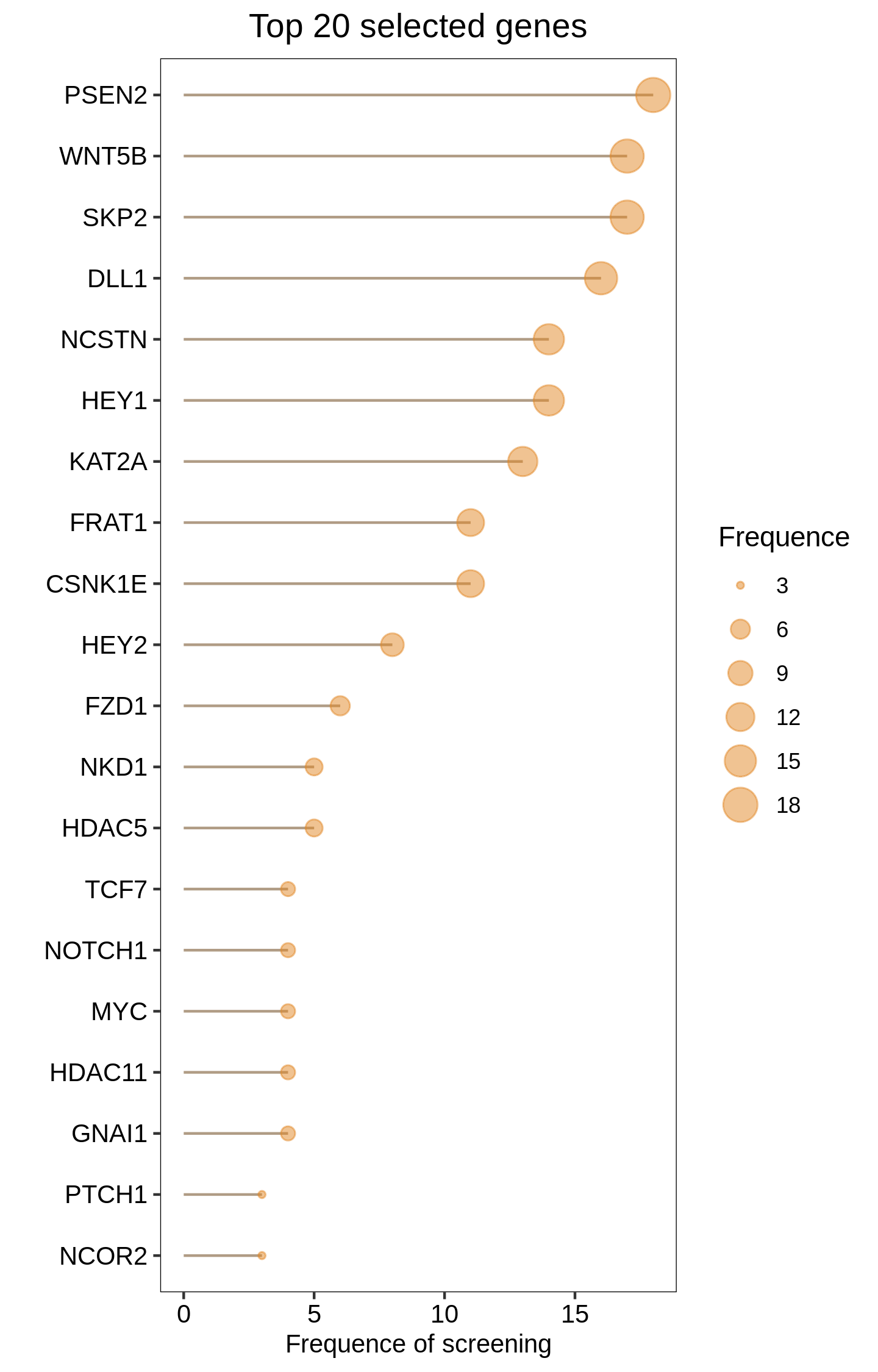
core_feature_rank(res.feature.all, top=20)
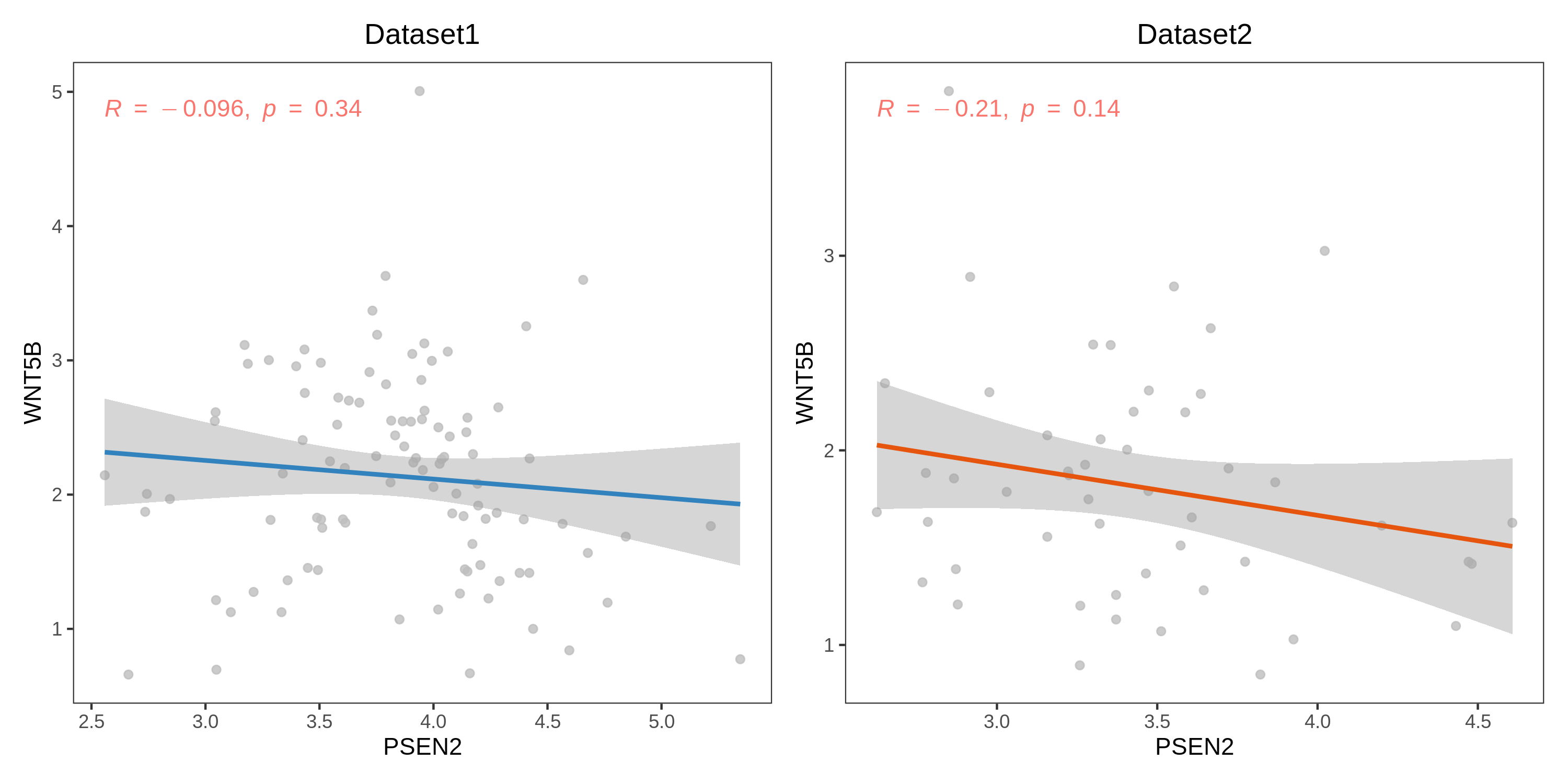
随机选择前两个基因来分析它们的相关性:
dataset_col<-c("#3182BDFF","#E6550DFF")
corplot <- list()
for (i in c(1:2)) {print(corplot[[i]]<-cor_plot(list_train_vali_Data[[i]],dataset=names(list_train_vali_Data)[i],color = dataset_col[i],feature1="PSEN2",feature2="WNT5B",method="pearson"))
}
aplot::plot_list(gglist=corplot,ncol=2)
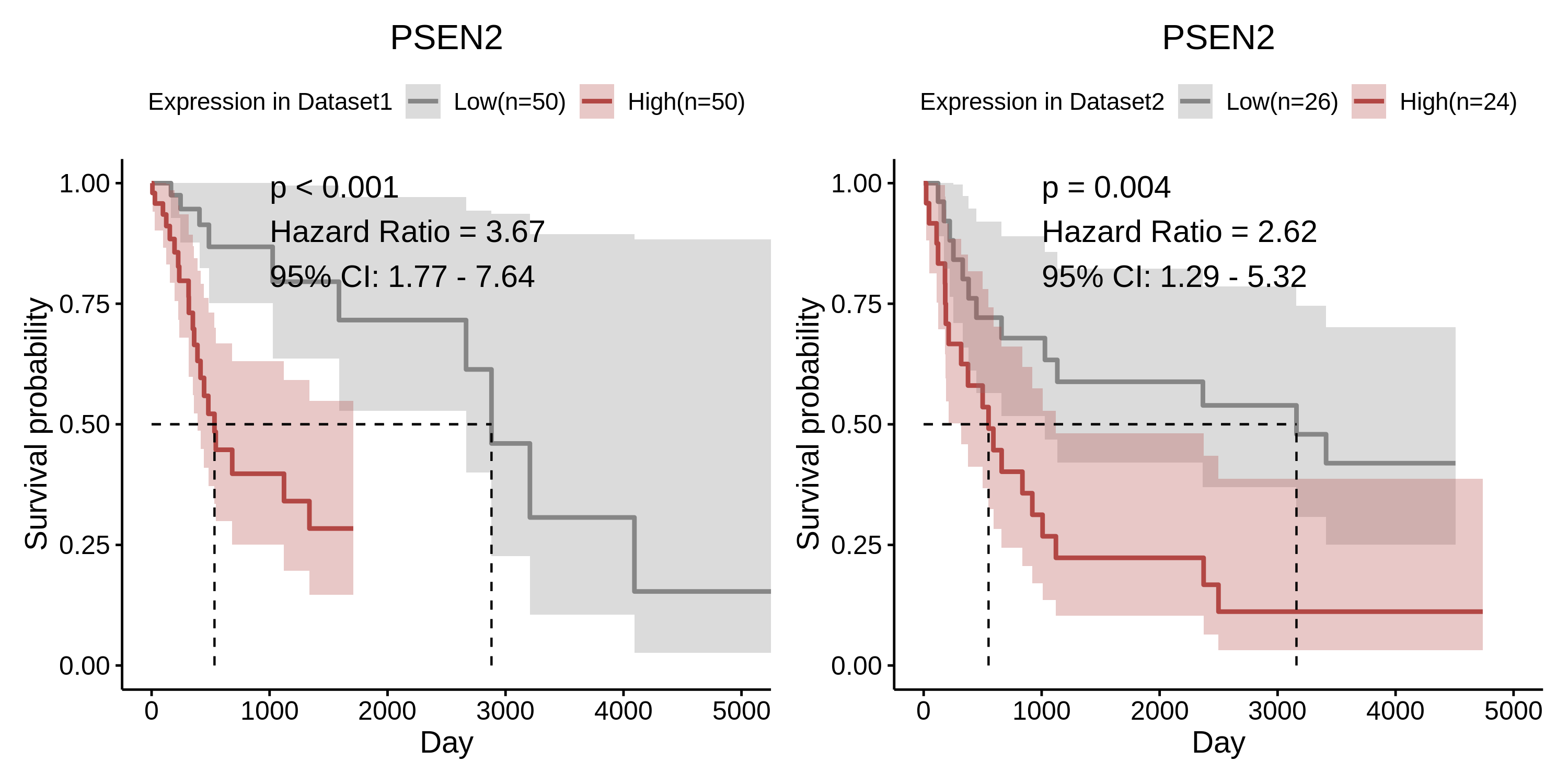
根据不同数据集中特定基因的中位表达水平绘制患者生存曲线:
survplot <- vector("list",2)
for (i in c(1:2)) {print(survplot[[i]]<-core_feature_sur("PSEN2", InputMatrix=list_train_vali_Data[[i]],dataset = names(list_train_vali_Data)[i],#color=c("blue","green"),median.line = "hv",cutoff = 0.5,conf.int = T,xlab="Day",pval.coord=c(1000,0.9)))
}
aplot::plot_list(gglist=survplot,ncol=2)
若我们的教程对你有所帮助,请点赞+收藏+转发,大家的支持是我们更新的动力!!
2024已离你我而去,2025加油!!
2024年推文汇总 (点击后访问)
2023年推文汇总 (点击后访问)
2022年推文汇总 (点击后访问)
往期部分文章
1. 最全WGCNA教程(替换数据即可出全部结果与图形)
- WGCNA分析代码六
推荐大家购买最新的教程,若是已经购买以前WGNCA教程的同学,可以在对应教程留言,即可获得最新的教程。(注:此教程也仅基于自己理解,不仅局限于此,难免有不恰当地方,请结合自己需求,进行改动。)
2. 精美图形绘制教程
- 精美图形绘制教程
- 《R语言绘图专栏–50+图形绘制教程》
3. 转录组分析教程
-
转录组上游分析教程[零基础]
-
一个转录组上游分析流程 | Hisat2-Stringtie
-
Samll RNA上游分析
4. 转录组下游分析
-
批量做差异分析及图形绘制 | 基于DESeq2差异分析
-
GO和KEGG富集分析
-
单基因GSEA富集分析
-
全基因集GSEA富集分析
BioinfoR生信筆記 ,注于分享生物信息学相关知识和R语言绘图教程。