【群体结构 ADMIXTURE之一】: fast ancestry estimation
原理:贝叶斯

ADMIXTURE
1 为什么要分析群体结构structure?
Structure软件分析达到的目的其实也很简单——分析群体的结构。要获知群体的结构,其实在前两篇与大家分享的两个图形也是可以解决的——PCA和树。通过树形图或PCA散点图,我们也可以直观了解个体间的分类关系(如图3)。
但如果需要解答:这个群体应该分为3个亚群还是4个亚群更合理,群体间是否存在基因交流(直白的说法:是否有杂交),以及每个个体混血程度是多少?对于PCA图和树形图,这解决不了,那么这时候我们就用到了structure图。
Structure图在开发之初,就使用了与进化树或PCA截然不同的算法,所以能够解决后两者不能解决的问题。具体的算法思路我在下文会解释,我们先直接解读structure图形的意义。当然,我们也不能完全不懂structure的基本原理就直接解读structure图。
PCA和树的不足,如上文提到的,无法推算:
1、群体应该划分为几个亚群;
2、群体间基因交流的程度;
3、某个个体的混血程度。
三、 祖先成分分析
祖先成分分析主要是探究个体的混血程度和群体间的基因交流程度。目前,可以做祖先成分分析的软件主要有 STRUCTURE,frqppe 和 admixture。 admixture运算速度快,操作简单,成为了主流的分析软件。
原理:Structure是由斯坦福大学Pritchard实验室开发的一款群体结构分析软件。structure可通过贝叶斯聚类方法对每个样本的来源进行判断,进而反应群体的遗传结构。真正跑时burnin设为100000,跑100000代,k设为1-20,每轮运行10次。
1. 简单介绍admixture算法原理
群体结构分析 - 斩毛毛 - 博客园
admixture用于从多位点SNP基因型数据集对个体祖先进行最大似然估计,虽然与STRUCTURE使用相同的统计模型,但其快速数值优化算法可以更快速地进行计算估计。具体来说,admixture使用 block relaxation 方法来交替更新等位基因频率和血统比例参数。通过解决大量独立凸优化问题来处理每个block更新,而这些凸优化问题则使用快速序列二次规划算法来解决。通常来说,SNP基因型数据集中的个体应该是不相关的,例如case-control相关性研究中的个体。
1、获得一系列样本的基因型,也就是变异信息 例如,SNP信息
2、在大部分情况下,我并不知道这个群体实际包含几个亚群。我们把群体的亚群数称为K值。那么我可以预设群体亚群数等于1~n,即K=1~n。那么软件就会模拟在K=x的情况下,使用贝叶斯算法推算群体是如何分群的,以及每个个体的血统构成是如何的。

独家 | 一文读懂贝叶斯分类算法(附学习资源) - 知乎
3、structure的计算原理
structure分析先预设群体由若干亚群(k=x)构成,通过模拟算法找出在k=x的情况下,最合理的样本分类方法。最后再根据每次模拟的最大似然值,找出最适用这群体的K值。
这个过程的逻辑如下:
1).亚群内符合哈温平衡
那么,软件在如何确定样本的最优分类方法呢?其实基于一个假设:在各个亚群内部个体应该符合哈代-温伯格平衡(哈温平衡的概念可以在百度查询),那么这个亚群内的基因频率分布应该可通过哈温平衡检验。例如,现在有40个个体的1个SNP位点的基因型,我预设亚群数k=2。我先随机将40个个体分成两份,然后检验是否符合哈温平衡。如果不符合,我继续调整分类策略,直到找到一种最优的分类方法:40个个体被分为了两份,每个亚群都由若干个体构成,每个亚群内部都最大程度地符合哈温平衡。
2).每个位点是独立的
同一个体基因组上的不同SNP可能来源不同亚群体,这是由于杂交混血过程带来的效应。如下图的D06个体,就各有部分DNA来自红色和黄色的亚群体。从另一个维度理解,为了达到哈温平衡,对不同的位点的分类方法是不同的。例如下图中,位点1的分类和位点2的分类策略就不同。位点1将D06个体划为红色亚群,而位点2将D06个体划分为黄色亚群。也就是说,软件是对每个位点单独进行分群的。

图 structure分析要求位点是独立的
3).每个样本的血统构成
既然对每个位点都完成分群了,自然最终就可以计算每个个体的血统构成了。
如果大家明白了这三个步骤了,我再以k=2为例,解释一下structure是如何找到样本的最优分类。其实简单说来,就是利用了计算机超强的运行能力,一开始计算机只是随机将样本分为两份,然后在每个亚群内进行哈温平衡检验。如果不符合哈温平衡(拍脑袋的分类,一开始当然是惨不忍睹),计算机继续调整分类,然后继续检验。
如此这般,在计算n次后,计算机再从这一堆结果中找到最佳的分类。这个过程称为“隐马科夫-蒙特卡罗链”的过程,计算次数n就是这个链的长度,这是structure一个重要的参数“Number of MCMC Reps”,需要预先设定。
但因为这个计算的过程是从随机模拟开始的。如果一开始拍脑袋拍的不好(随机分类与真实分类差距太大),计算机一黑到底,最后把n次用完了,都没有找到一个合理的分类。所以,分析软件往往有个预实验的过程。
就是在正式进行大规模运算前,计算机先尝试各种各样的随机分类,运行非常短的次数,然后评估哪种随机分类是最合理的。之后,在根据最优的随机分类,进行后续的大规模运算。这个过程就称为burn-in period,预实验的次数就称为burin-in的次数。这也是structure分析另外一个重要的参数“length of burn-in period”。
如果你理解了这两个参数ok,非常好。Structure软件最重要的两个参数你已经掌握了。剩下就选择使用那种模型了。主要涉及两种模型 no admixture model和admixture model。前者假设亚群间不存在杂交,后者则假设亚群间存在杂交。在绝大部分情况下,当然是选择admixture 模型更合理了。群体结构分析structure原理 - 组学大讲堂问答社区
第三步:寻找合适的K值
-
for i in {1..7};do admixture --cv test1.bed $i |tee log${i}.out;done ## 根据CV error 确定K值 grep -h 'CV' log*.out CV error (K=1): 0.22317 CV error (K=2): 0.15018 CV error (K=3): 0.12804 CV error (K=4): 0.12109 CV error (K=5): 0.12656
当k=4时,cv error 最小,选择4
此外,
如果SNP数据集非常大,则可以随机选择SNP进行K值选择分析,比如随机选取20000个SNP进行分析,每个K值跑20次,确定最终的k值,然后分析
2. 数据处理和祖先成分估计
-
数据过滤
#!/bin/bashplink=/software/biosoft/software/plink/plinktime $plink --vcf 60_dog.merge.vcf --dog --geno 0.05 --maf 0.05 --hwe 0.0001 --make-bed --out 60_dog_qc && echo "----QC and vcf2bed done"
-
估计最优祖先个数,即群体分为几个亚群更合理,这里亚群数目称为K。通常情况下,由于不知道这个群体实际包含几个亚群,可以预设K的范围为2~n。软件就会模拟在K=x的情况下,推算群体是如何分群的,以及每个个个体的血统构成比例。对每个K值模拟的结果,软件都会计算出一个CV error值和最大似然值,error值和最大似然值两个指标都可以挑选最佳K值。对于error值来说
admixture=/software/biosoft/software/admixture_linux-1.3.0/admixturefor K in {2..5}
dotime $admixture --cv 60_dog_qc.bed $K |tee log${K}.out && echo "----admixture K=$K done----"
done
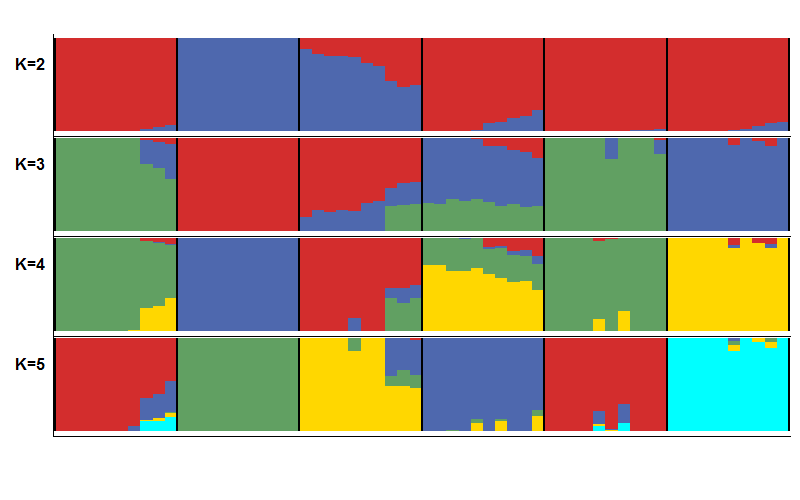
对每个K值模拟的结果,软件都会计算出一个CV error值和最大似然值,error值越小越好,似然值越大越好,两个指标都可以挑选最佳K值。一般而言就是要挑选出error值最小和K值最小的分群策略或者挑选出似然值最大和K值最小的分群策略,即"拐点"。然而,目前学界对于如何判断分群最佳仍有很大争议,CV error值和最大似然值只是参考,无法起决定性作用,因为,目前主流做法是把K值(2~n)都画出来,分别解释。这里取K=2~5,来看下具体结果。

很不幸,这里error值和似然值都是单调的,没有所谓的拐点,可以都画出来看一下。
-
admixture运行结束后,会产生.Q文件,记录每个个体不同群体祖先成分信息,以K=2为例截图看下

添加上样本ID和所属分组ID,进行可视化。这里以K=2时,来看下处理后的文件

3. 祖先成分堆叠图可视化
这里使用R完成祖先成分堆叠图可视化,由于篇幅限制,这里只放主体部分
所以K=2时,堆叠图是这样的

按照上述代码,依次画出k=2~5的堆叠图p2,p3,p4,p5,并使用R包cowplot 组合在一起,代码如下
combine_bar<-ggdraw() + draw_plot(p2,0.05,0.7,0.95,0.25) + draw_plot(p3,0.05,0.5,0.95,0.25) + draw_plot(p4,0.05,0.3,0.95,0.25) + draw_plot(p5,0.05,0.1,0.95,0.25) + draw_plot_label(c("K=2", "K=3", "K=4","K=5"), c(0, 0, 0, 0), c(0.9, 0.7, 0.5,0.3), size = 12)
combine_bar
合并之后,可视化就基本完成了。
以上参考了比较多的博主文章
admixture 群体结构分析
群体结构分析三种常用方法(下篇)
【R语言】群体结构可视化R语言包pophelper的使用
Admixture使用说明文档cookbook - 知乎
admixture 学员来稿|全基因组关联分析(GWAS)学习笔记分享(三)_搜狐汽车_搜狐网