日拱一卒 | RNA-seq数据质控(1)
拿到测序得到的RNA-seq的fastq数据之后,首先需要对测序数据进行质控,检查一下数据的质量。那么如何进行质控呢?
之前我对这个的理解总是云里雾里的,现在想对此认真地理一理。也希望我的学习结果可以分享给更多的人。
一、碱基质量表征
我们拿到fastq数据之后,需要评估每一个位点测出来的碱基是这个碱基的可靠性。而在这里,测序平台在返还的fastq的数据中就已经包含了这部分信息。我们要做的就是利用这些信息,对序列进行过滤和修剪,保留高质量的测序读段,便于下游的比对等环节。
1.经典的fastq序列的结构
经典的fastq序列由以下四部分组成:@开头的序列名;我们测到的碱基序列;+;一些近似于乱码的符号。
这里有用的部分就是第二行和第四行的信息。第二行是测序平台测到的序列,而第四行则每一个符号和第二行的碱基一一对应,表示的是碱基的测序质量。为什么这些乱码的符号可以表示碱基的测序质量呢?这里存在有一个转换。
@LH00308:393:22TCC7LT4:1:1101:41256:1028 2:N:0:CTCCGTTA+TNGGAACC
NATACCGTGGCATTGACTGCCTTGCTGTGGCCTATGGCGTAGCTGTTAACAAGCTCACACTGCCCATCCCATCCACCTGCCCCGAGCCCTTCGCACAGCTTATGGCCGACTGCTGGGCGCAGGACCCCCACCGCANGNNNGACTTCGCNT
+
#IIIIIIIIII9IIIIIIIIIIIIIIIIIII*IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII#I###IIIIIIII#I
2.碱基的测序质量表征
公式如下,其中p为测序出错率,比如可以为0.01,即该碱基测错的可能性为1/100。p越小,说明碱基的质量越高;相应地,经过下面的公式转换后,计算得到q为20。上述转换即把测序质量进行更加直观的表示,q越大,说明测序质量越高;q的取值范围在0-40之间;并且这样转换的另一个好处是,可以将q的数值和ASCII的字符编码一一对应起来(为了对应方便会加一个33,ASCII的可见字符分布在33-126之间),从而实现单个碱基和表征测序质量的单个字符一一对应,方便计算机存储。这个转换可以说非常的巧妙。
q = − 10 ∗ l o g 10 ( p ) q=-10*log_{10}(p) q=−10∗log10(p)
下图表示的是ASCII码和数值的对应关系。

那么借此,我们就可以对fastq文件中序列的测序质量进行查看和分析。
二、碱基质量在质控结果中的体现
我们一般常用fastQC来对fastq文件进行质量检查(代码实现环节此不赘述),其输出了许多的指标,其中有关碱基质量的有如下:
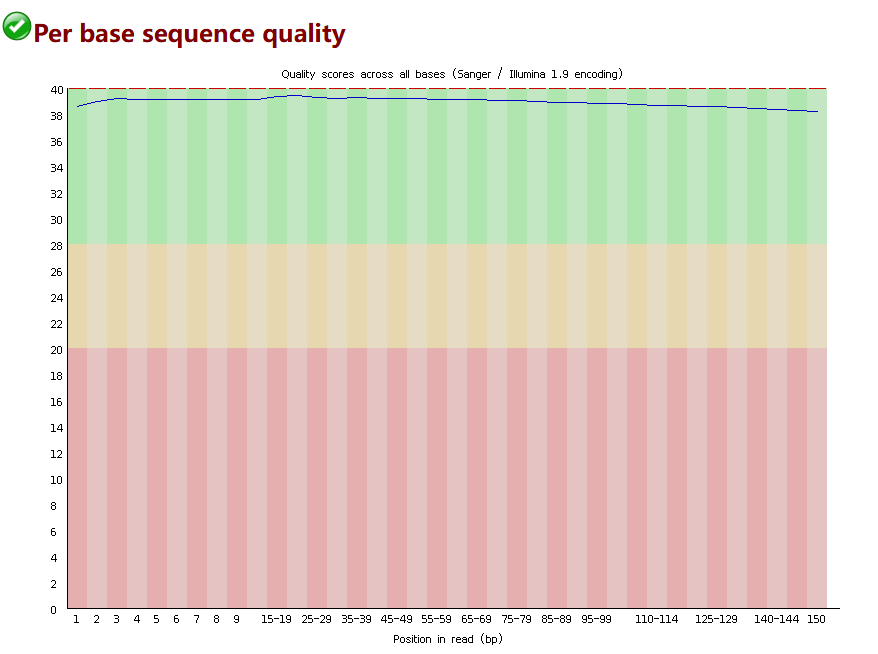
1. 读段每一个碱基位置的碱基质量分布
通常以下图的形式进行展示(下面这张图算是质量比较好的)。其中行,为读段的位置,我的数据是经典的150bp。列为碱基的质量,即前面提到的q(在0-40之间)。这张图表示的是,对于这个文件中的读段,分别统计每一个位置的碱基质量,然后绘制boxplot。每一个位点有一个boxplot。那么可以得知测序的过程中,测到的碱基质量是否具有位置的偏好性(其中一种常见的情况是,在读段的后半部分,碱基质量会降低,这提示我们可能需要根据位置进行修剪)。
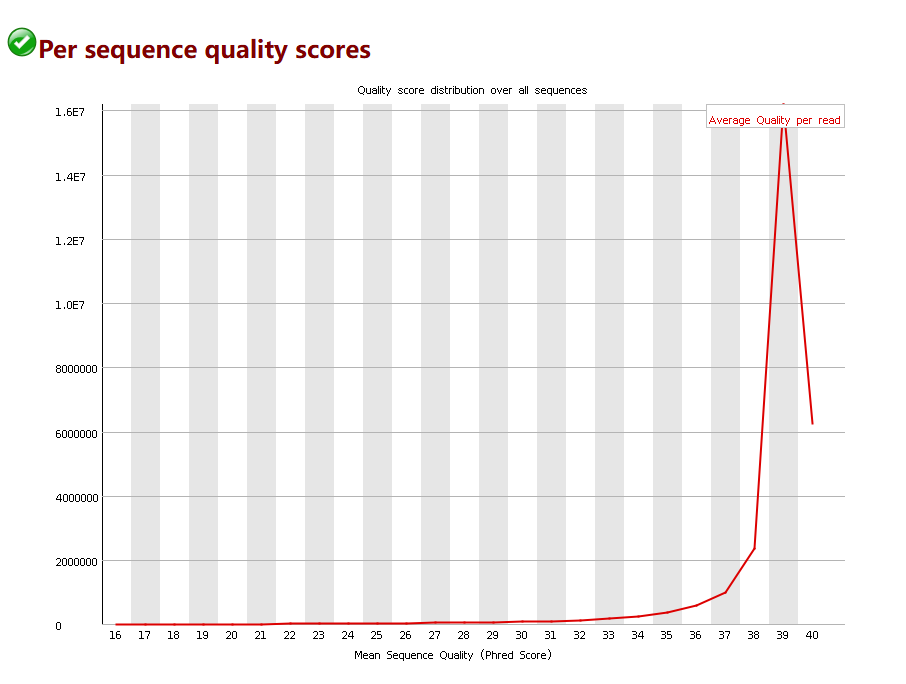
2. 所有读段的平均碱基质量
我们还可以统计每一个读段的平均碱基质量,绘制碱基质量的分布曲线,从而可以对质量较差的读段进行整个的过滤。下图显示大部分的读段的平均质量在39附近。
三、针对于碱基质量的处理
主要有两个思路,第一,对于整体质量较低的读段,直接按照质量的阈值进行过滤。第二,对于可能具有位置偏好性的读段(比如前面或者后面的测序质量很差),则可以针对位置对读段进行修剪,保留高质量的读段片段。
这里需要注意的一点是,对于双端测序的读段序列,可能存在一对中的其中一个不符合质控条件而被剔除。由于后续的比对环节通常要求双端测序的文件在序列顺序上是一致的,因此需要使用在处理的时候考虑将双端测序文件在处理后依然保持一致的工具,比如trimmomatic。