Tangram官网教程
分析
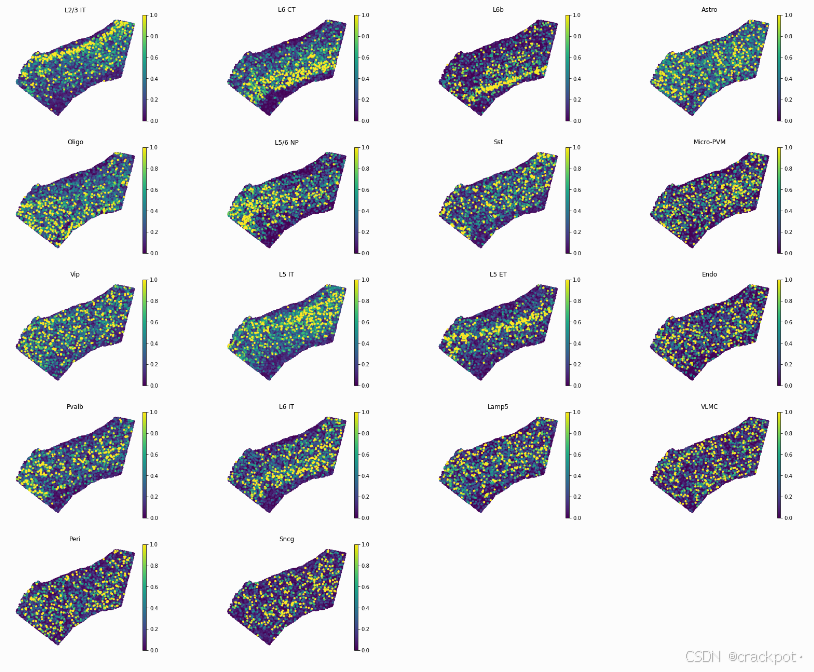
将单细胞数据映射到空间上最常见的应用是把细胞类型注释转移到空间层面。 这是通过`plot_cell_annotation`函数实现的,它能将`obs`数据框中的注释(此处为`subclass_label`字段)以空间概率图的形式可视化。你可以将`robust`参数设置为`True`,同时设置`perc`参数来确定颜色映射的范围,这有助于去除异常值。 下面的图恢复了兴奋性神经元的皮质层以及神经胶质细胞的稀疏分布模式。正如预期的那样,皮质的边界由6b层(细胞类型为L6b)界定,并且少突胶质细胞集中分布在皮质下区域。 然而,VLMC细胞类型的分布模式似乎不太对:VLMC细胞通常聚集在皮质第一层,但在感兴趣区域(ROI)却很稀疏。这通常意味着:(1)我们在训练基因中没有为VLMC细胞选择合适的标记基因;(2)现有的标记基因在空间数据中非常稀疏,因此无法提供良好的映射信号。
tg.project_cell_annotations(ad_map, ad_sp, annotation='subclass_label')
annotation_list = list(pd.unique(ad_sc.obs['subclass_label']))
tg.plot_cell_annotation_sc(ad_sp, annotation_list, x='x', y='y', spot_size=60, scale_factor=0.1, perc=0.001)- 使用
Tangram的project_cell_annotations函数将单细胞数据的subclass_label注释投影到空间转录组数据上。 - 获取单细胞数据中
subclass_label列的唯一值列表,使用Tangram的plot_cell_annotation_sc函数绘制细胞注释的空间分布。
让我们尝试更深入地了解这种映射的效果有多好。`plot_training_scores`函数能很好地帮助我们实现这一点,它会生成四个图表: 第一个图表是每个训练基因相似度得分的直方图。大多数基因的映射相似度非常高(> 0.9),但也有少数基因的得分在0.5左右。我们想弄清楚为什么这些基因的得分较低。 第二个图表展示了基因的训练得分(y轴)与该基因在单细胞核RNA测序(snRNA - seq)数据中的稀疏性(x轴)之间存在明显的相关性。每个点代表一个训练基因。趋势是基因越稀疏,得分越高:出现这种情况通常是因为非常稀疏的基因更容易映射,因为只需在正确的位置放置几个 “关键细胞” 就能匹配它们的模式。 第三个图表与第二个类似,但展示的是空间数据中的基因稀疏性。空间数据通常比单细胞数据更稀疏,这种差异往往是导致映射质量较低的原因。 在最后一个图表中,我们展示了训练得分与两个数据集之间稀疏性差异的函数关系。对于稀疏性相当的基因,映射得到的基因表达与空间数据中的基因表达非常相似。然而,如果一个基因在一个数据集中(通常是空间数据集)非常稀疏,而在另一个数据集中不稀疏,那么映射得分就会较低。这是因为由于两个数据集之间的缺失值数量不一致,Tangram无法正确匹配基因模式。
tg.plot_training_scores(ad_map, bins=10, alpha=.5)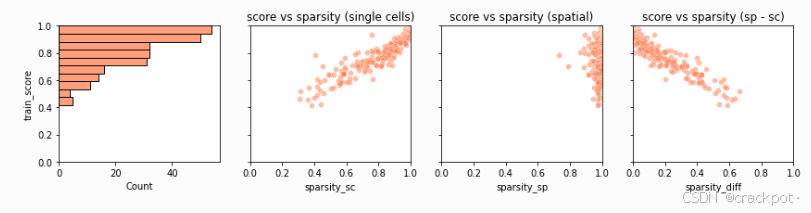
尽管上述图表为我们提供了单基因水平的得分汇总,但我们还需要知道哪些基因的映射得分较低。 这些信息可以从映射结果的`adata.uns['train_genes_df']`数据框中获取;该数据框就是用于绘制上述四个图表的数据框。 我们想要检查映射得分较低的训练基因的表达情况,以此了解映射的质量。 首先,我们需要利用映射后的单细胞生成 “新的空间数据”:这可以通过`project_genes`函数来完成。 该函数接受映射结果(`adata_map`)和相应的单细胞数据(`adata_sc`)作为输入。 其结果是一个体素(voxel)-基因形式的AnnData对象,形式上与`ad_sp`类似,但包含的是映射后的单细胞数据的基因表达,而非Slide-seq数据的基因表达。
ad_ge = tg.project_genes(adata_map=ad_map, adata_sc=ad_sc)
ad_ge
AnnData object with n_obs × n_vars = 9852 × 26496obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'x', 'y', 'uniform_density', 'rna_count_based_density'var: 'n_cells', 'sparsity', 'is_training'uns: 'training_genes', 'overlap_genes'我们现在选择一些映射得分较低的训练基因。
genes = ['rgs6', 'satb2', 'cdh12']
ad_map.uns['train_genes_df'].loc[genes][19]:
| train_score | sparsity_sc | sparsity_sp | sparsity_diff | |
|---|---|---|---|---|
| rgs6 | 0.462164 | 0.305172 | 0.941941 | 0.636769 |
| satb2 | 0.501113 | 0.455904 | 0.969549 | 0.513645 |
| cdh12 | 0.417188 | 0.384889 | 0.972594 | 0.587705 |
为了可视化基因模式,我们使用辅助函数`plot_genes`。这个函数接受两个体素 - 基因格式的AnnData对象作为输入:实际的空间数据(`adata_measured`)和Tangram的空间预测数据(`adata_predicted`)。该函数会返回这两个空间AnnData对象中指定基因`genes`的基因表达图谱。 正如预期的那样,尽管预测的基因表达捕捉到了主要模式,但稀疏程度更低。对于这些基因,我们更相信映射得到的基因模式,因为Tangram通过在空间上对齐稀疏度较低的数据来 “校正” 基因表达。
tg.plot_genes_sc(genes, adata_measured=ad_sp, adata_predicted=ad_ge, spot_size=50,scale_factor=0.1, perc=0.02, return_figure=False)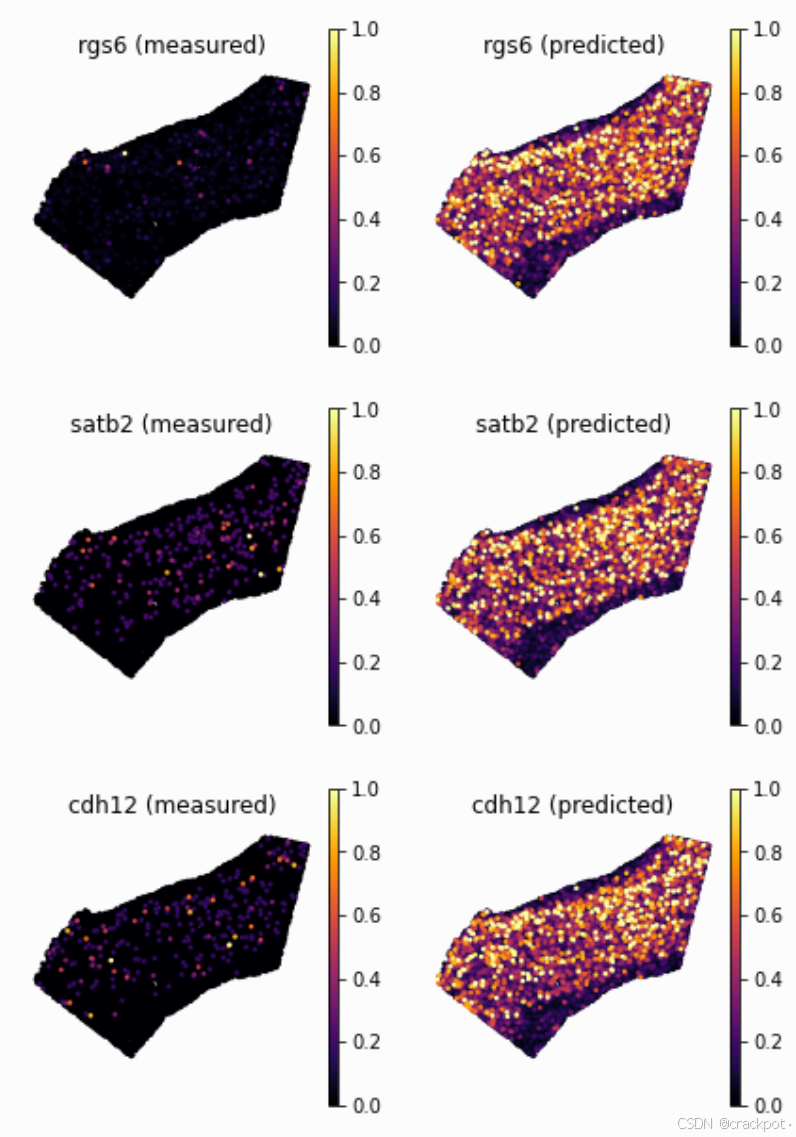
在那些在空间数据中未被检测到,但在单细胞数据中能被检测到的基因上,我们能看到更具说服力的例子。在使用`pp_adatas`函数进行训练之前,这些基因就被剔除了。不过,Tangram仍能就这些基因的空间模式可能是什么样给出见解。
genes=['mrgprx2', 'muc20', 'chrna2']
tg.plot_genes_sc(genes, adata_measured=ad_sp, adata_predicted=ad_ge, spot_size=50, scale_factor=0.1, perc=0.02, return_figure=False)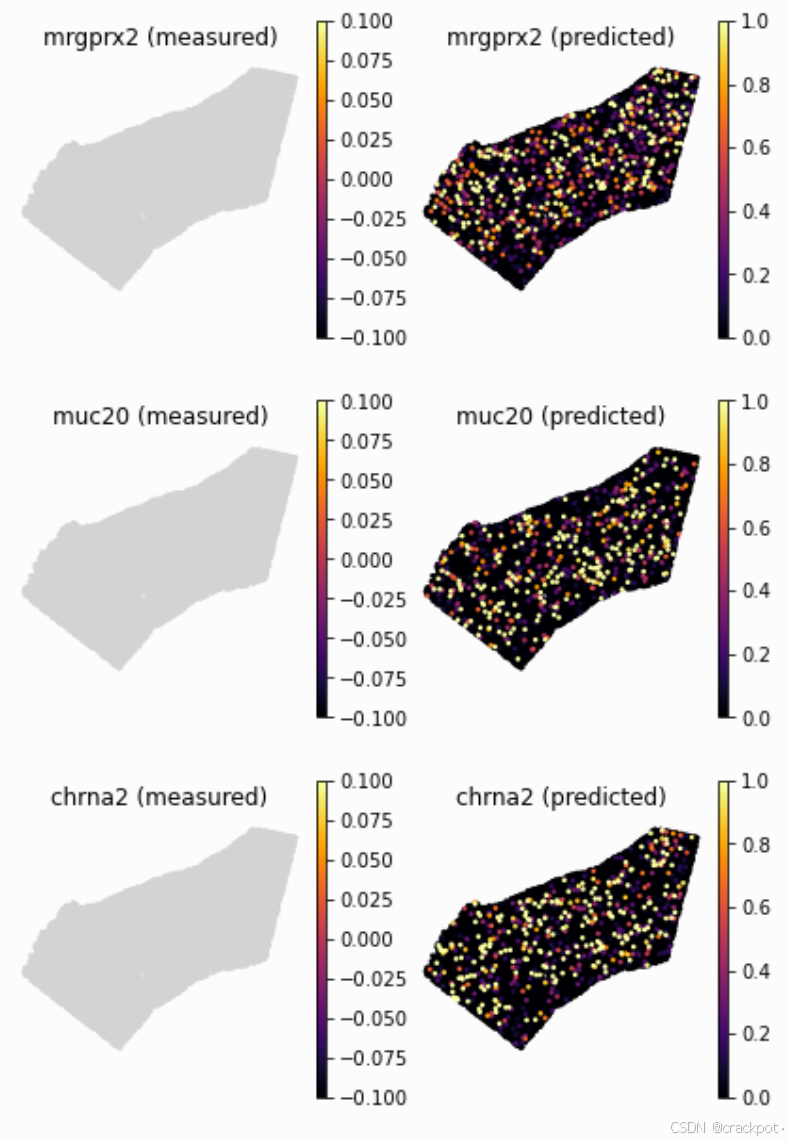
到目前为止,我们只检查了用于数据比对的基因(训练基因),但是经过映射的单细胞数据`ad_ge`包含了整个转录组信息,其中包括超过2.6万个测试基因。
(ad_ge.var.is_training == False).sum()[22]:26247
我们可以使用`plot_genes`函数来检查非训练基因的表达情况。由于基因表达预测是验证映射效果的方式,所以这是一个关键步骤。 在进行此操作之前,计算所有基因的相似度得分会很有帮助,这可以通过`compare_spatial_geneexp`函数完成。该函数接受两个空间`AnnData`对象(即体素-基因格式的数据),并返回一个包含所有基因相似度得分的数据框。训练基因通过布尔字段`is_training`进行标记。 如果我们像下面这样将单细胞`AnnData`对象也传递给`compare_spatial_geneexp`函数,返回的数据框将包含额外的稀疏性列——`sparsity_sc`(单细胞数据的稀疏性)和`sparsity_diff`(空间数据稀疏性与单细胞数据稀疏性的差值)。如果我们之后想用`compare_spatial_geneexp`函数返回的数据框来调用`plot_test_scores`函数,这一步是必要的。
df_all_genes = tg.compare_spatial_geneexp(ad_ge, ad_sp, ad_sc)
df_all_genes| score | is_training | sparsity_sp | sparsity_sc | sparsity_diff | |
|---|---|---|---|---|---|
| igf2 | 9.996735e-01 | True | 0.994011 | 0.999924 | -0.005913 |
| chodl | 9.967118e-01 | True | 0.999086 | 0.999016 | 0.000070 |
| 5031425f14rik | 9.963596e-01 | True | 0.999594 | 0.998789 | 0.000805 |
| car3 | 9.943125e-01 | True | 0.999695 | 0.999016 | 0.000679 |
| scgn | 9.935710e-01 | True | 0.999898 | 0.999205 | 0.000693 |
| ... | ... | ... | ... | ... | ... |
| gm3376 | 1.477303e-08 | False | 0.999898 | 0.999962 | -0.000064 |
| gm21317 | 1.057379e-08 | False | 0.999898 | 0.999962 | -0.000064 |
| sprr2d | 9.872679e-09 | False | 0.999898 | 0.999962 | -0.000064 |
| cd69 | 7.458404e-09 | False | 0.999898 | 0.999962 | -0.000064 |
| cyp1a2 | 7.139468e-09 | False | 0.999898 | 0.999962 | -0.000064 |
18000 rows × 5 columns
下面的图表为我们提供了测试基因在单基因水平上的得分汇总情况。
tg.plot_auc(df_all_genes)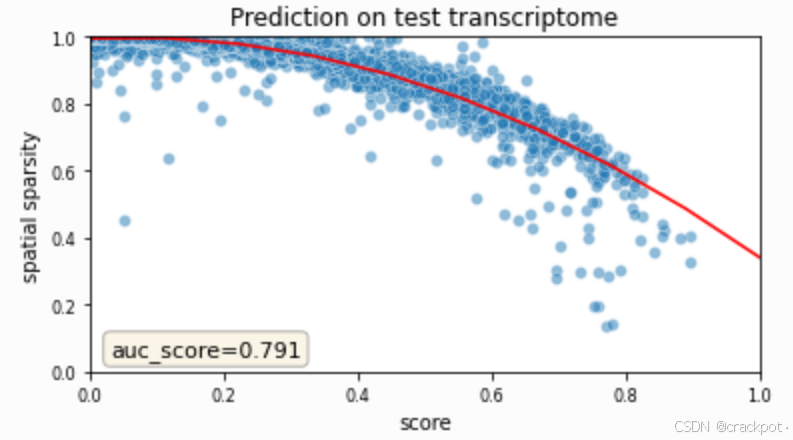
让我们绘制测试基因的得分图,看看它们与训练基因相比情况如何。沿用之前绘图的策略,我们将得分作为空间数据稀疏性的函数进行可视化。 (我们还没有将这个调用封装成一个函数。) 同样地,空间数据中更稀疏的基因预测得分较低,这是由于空间数据中存在数据缺失的情况。 让我们挑选几个具有不同得分的测试基因,对比一下预测的基因表达和实际测量的基因表达。
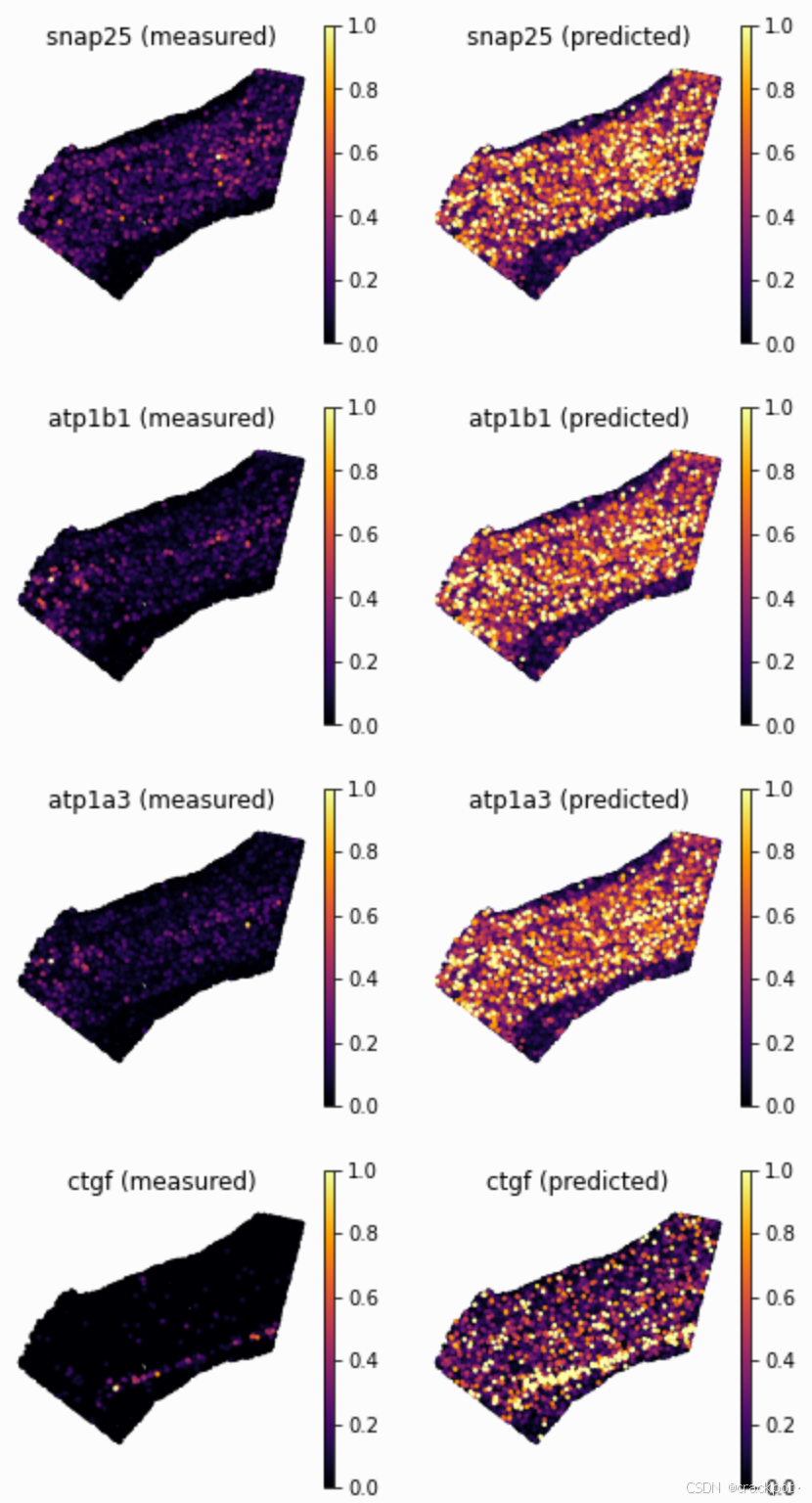
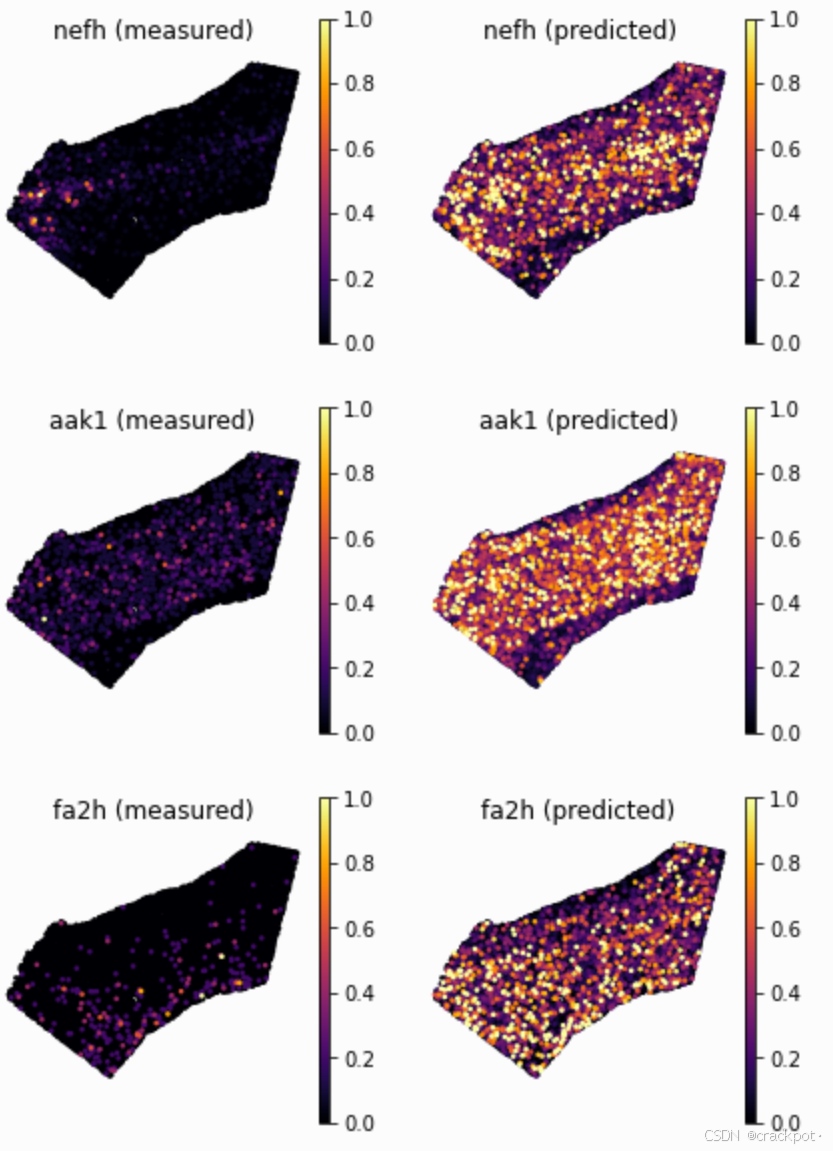
genes = ['snap25', 'atp1b1', 'atp1a3', 'ctgf', 'nefh', 'aak1', 'fa2h', ]
df_all_genes.loc[genes]| score | is_training | sparsity_sp | sparsity_sc | sparsity_diff | |
|---|---|---|---|---|---|
| snap25 | 0.897492 | False | 0.402253 | 0.120048 | 0.282205 |
| atp1b1 | 0.824424 | False | 0.579984 | 0.175778 | 0.404205 |
| atp1a3 | 0.753856 | False | 0.658343 | 0.319587 | 0.338757 |
| ctgf | 0.585824 | False | 0.981628 | 0.948243 | 0.033386 |
| nefh | 0.536002 | False | 0.909156 | 0.916083 | -0.006928 |
| aak1 | 0.538055 | False | 0.868047 | 0.179713 | 0.688334 |
| fa2h | 0.363725 | False | 0.972493 | 0.860845 | 0.111648 |
我们可以再次使用 `plot_genes` 函数来可视化基因模式。 有趣的是,尽管基因 `Atp1b1` 或 `Apt1a3` 的得分更高,但它们与实际情况的吻合度似乎不如 `Ctgf` 和 `Nefh`。这是因为,尽管后两个基因的模式定位正确,但其表达值的相关性却不是很好(例如,在 `Ctgf` 基因中,“亮黄色区域” 位于 6b 层的不同位置)。相比之下,对于 `Atpb1` 基因,尽管空间数据中的整体基因表达更微弱,但基因表达模式在很大程度上得到了恢复。
tg.plot_genes_sc(genes, adata_measured=ad_sp, adata_predicted=ad_ge, spot_size=50,scale_factor=0.1, perc=0.02, return_figure=False)