红花多组学挖掘OGT1-文献精读146
Genome-wide screen and multi-omics analysis reveal OGT1 participate in the biosynthesis of safflower flavonoid glycosides
基因组范围筛选和多组学分析揭示OGT1参与了红花黄酮苷类化合物的生物合成

摘要
红花作为一种经济作物,以其花朵闻名,广泛用于治疗心血管和脑血管疾病的药物中,以及食品和工业染料中。红花的药用价值依赖于其黄酮苷类化合物。因此,红花黄酮苷类化合物的生物合成一直是关注的重点,但目前的机制仍然不甚明确。本研究旨在通过整合全基因组筛选和多组学关联研究的综合方法,鉴定与红花黄酮苷类化合物生物合成相关的功能基因。CYP和UGT是参与黄酮苷类化合物生物合成的两个关键基因家族。我们筛选了红花基因组中的264个CYP基因和140个UGT基因,并进行了包括系统发育关系、保守基序、基因结构、顺式作用元件和染色体定位等分析,为CYP和UGT基因家族提供了广泛和全面的数据。通过整合来自红花不同组织的表型和代谢数据,有助于通过确认HSYA仅在花朵中合成,进一步缩小筛选范围。基于基因表达模式和系统发育分析,最终确定了CtOGT1基因,该基因能够催化使用不同黄酮底物合成黄酮苷,并表现出强烈的底物亲和力。此外,分子对接研究阐明了CtOGT1的高度活跃内在机制。总之,本研究通过整合全基因组筛选和多组学分析,有效地鉴定了红花黄酮苷类化合物生物合成的相关基因,为进一步阐明红花黄酮苷类化合物生物合成途径建立了全面的数据、方法学和实验依据。
引言
红花(C. tinctorius L.),属于菊科,是一种有着超过4500年栽培历史的作物,作为一种经济作物具有重要的价值。它分布在多个国家,包括美国、中国、墨西哥、日本、印度和埃及[1]。红花花朵中含有多种黄酮苷类化合物。研究表明,这些化合物在治疗心血管和脑血管疾病方面具有显著的治疗效果。例如,羟红花黄A(HSYA)可以缓解脑缺血-再灌注损伤、血栓形成和动脉硬化等问题。红花作为药用植物在许多国家和地区得到广泛应用。在中国,它是超过300种传统方剂的主要成分之一,包括丹红注射液,用于治疗中风、冠心病和心绞痛[2, 3]。在日本,红花是汉方药物的必备成分,用于改善血液循环和减少血瘀。此外,红花的黄酮提取物被用作健康的天然色素。在古埃及、日本和印度,红花衍生的黄酮类化合物在传统服装的染色中起着重要作用。这些色素还被用作天然食品染料,如面包、蛋糕、饼干、糖果和饮料[4]。在工业上,它们也用于药品和化妆品的着色[5]。
红花中的黄酮苷类化合物可以分为两大类:一类是大多数植物中常见的黄酮类和黄烷醇类,另一类是挑战素及其苷类,包括HSYA、羟红花黄B和红花苷A,这些也被称为红花特有的有色成分[6]。黄酮类和黄烷醇类的生物合成途径与模型植物和作物相似,且其生物合成途径已被阐明。许多关于这些黄酮化合物生物合成途径中共享基因的报告,例如CHS[7]、CHI[8]、F3H[9]和F3'5'H[10]。然而,这些特有成分,特别是黄酮苷类化合物,在红花中的生物合成途径仍不清楚。根据这些化合物的化学结构,它们的生物合成被认为是通过基于挑战素的糖基化和羟基化过程(图S1)。近年来,我们的研究重点是探讨这些成分的生物合成途径,如Ren等[11]筛选了糖基转移酶基因UGT3,Chen等[12]在红花中筛选了CtCGT1。同时,许多研究者也致力于阐明红花中黄酮苷类化合物的生物合成途径。例如,Xu等[13]克隆并确认了糖基转移酶(GT)基因在苷类化合物生物合成中的作用,特别是双功能GT。Wang等[14]鉴定并验证了红花中的新型细胞色素氧化酶基因。然而,目前的基因筛选主要参考了其他物种中报告的基因,缺乏针对红花的全面全基因组视角。这种方法在阐明红花黄酮苷类化合物的生物合成途径方面提供的信息有限,仍然无法清楚揭示生物合成途径。
目前,功能基因的发现主要通过RNA-seq结合代谢组学进行差异基因筛选,以及基于基因组数据的同源比较和基因簇挖掘等策略[15]。在植物天然产物生物合成中,基因表达水平的提高通常与生物活性物质的积累呈正相关。转录组学和代谢组学追踪基因表达和物质积累在不同组织、发育阶段和环境压力下的变化。整合转录组学和代谢组学是研究植物发育背后的调控网络和生理机制的重要途径[16],并广泛应用于许多经济作物的研究。例如,Li等[17]整合了番茄不同组织和生长阶段的时空代谢组学和转录组学数据,构建了番茄的代谢调控网络;Wu等[18]对未成熟和成熟的黑莓果实进行了联合转录组学和代谢组学分析,以解密黄酮类化合物的生物合成机制。然而,这种方法通常止步于筛选差异表达基因或差异代谢物,在阐明特定分子机制方面仍然存在理论性。基于基因组数据的基因筛选通常旨在阐明特定基因的功能[19]。然而,用于筛选功能基因的同源比较存在许多局限性,往往需要以相关物种或同科、同属植物的功能基因作为参考。例如,Zhao等[20]通过比较四种唇形科植物的基因筛选了黄芩中的功能基因,Hagel等[21]也利用同科植物的基因筛选了紫草科植物中马克斯芭胶的功能基因。全基因组包含了丰富的基因家族信息,结合多组学分析可以有效且高效地用于基因组与表型关联及基因鉴定[22]。这种方法在小麦、油菜[23]和大豆[24]等多种经济作物中已被证明是有效的。
本研究旨在通过 cataloging红花基因组中CYP和UGT超级家族的所有成员,获取其系统发育、保守基序、基因结构和启动子顺式作用元件的详细信息,进而确定与黄酮苷类化合物生物合成相关的核心基因。根据红花根、茎、叶和花中疑似为HSYA的红色物质的存在,进行了表型-代谢物关联分析。对红花不同发育阶段、组织、光照强度和甲基茉莉酸(MeJA)处理下的转录组分析,探讨了这些因素对红花黄酮苷类化合物积累的影响。通过整合全基因组和多组学关联分析,筛选了参与红花黄酮苷类化合物生物合成的候选基因,并验证了其活性。总体而言,本研究关联了参与红花黄酮苷类化合物生物合成过程的关键基因家族、不同组织的表型和代谢特征以及各种条件下的转录组学结果,为黄酮苷类化合物生物合成功能基因的筛选提供了全面的策略,并为阐明其他植物生物活性成分的生物合成途径提供了方法支持。
结果
红花基因组中CYPs和UGTs的鉴定
红花的基因组数据来自我们之前的研究,且已经完成了基因注释[12]。我们根据Pfam注释检索了所有相关的序列。CYPs通过Pfam域PF00067进行特征化,UGTs通过Pfam域PF00201进行特征化。在红花中,我们共鉴定了264个CYP家族转录本和140个UGT家族转录本。
CYP的蛋白质长度从303到612个氨基酸不等,分子量介于34.08至69.68 kDa之间,理论等电点范围从5.42到10.15。亚细胞定位预测表明,大多数CYP蛋白(264个中的260个)位于内膜系统中。对于UGTs,蛋白质的长度变化范围为303到612个氨基酸,等电点从4.64到9.45,分子量从22.30到62.40 kDa。亚细胞定位显示,大部分UGT蛋白主要位于叶绿体(140个中的72个),其次最多的群体位于细胞膜上(140个中的19个)。CYPs和UGTs的详细特征,包括不稳定指数、脂肪指数和水合性总平均值,见表S1和S2。
红花中CYPs和UGTs的系统发育分析
为了进一步探讨红花中CYPs和UGTs的进化关系,我们分别基于修剪后的对齐构建了一个无根的最大似然(ML)树。此分析使得我们能够将264个CYP和140个UGT蛋白序列分配到特定的族群或组别(图1,详细信息见表S3)。
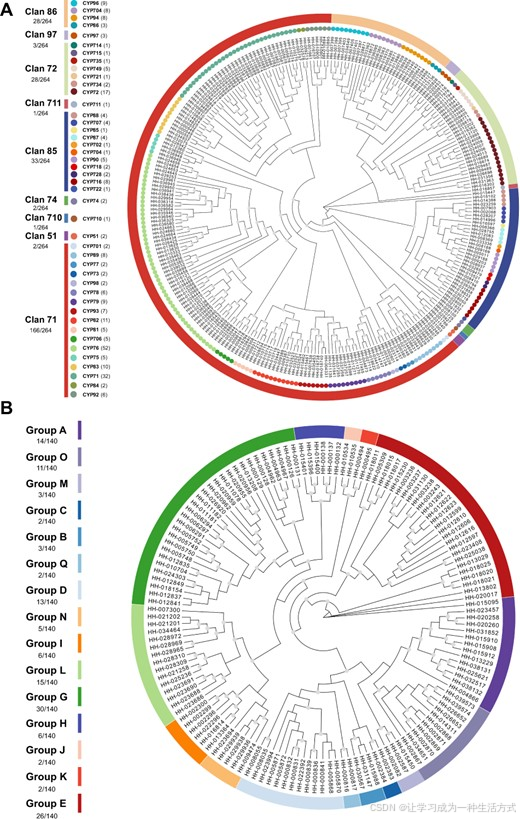
图1 红花中CYPs和UGTs的系统发育分析 为了构建最大似然树,使用了CYPs和UGTs的蛋白质序列,分析通过5000次自助法重复支持。自助法值大于0.7的分支,在每个分支的中心标记有灰色圆圈。CYP的族群和家族分别通过颜色条和圆圈进行区分(A),而UGT的各组则使用颜色条标记(B)。
在红花中,我们鉴定出了九个CYP族群,涵盖了总共45个家族。这些族群根据其在系统发育树上的位置被分为A型和非A型。A型仅包含71族群,这是一个多家族族群,包含17个家族,共计166个基因。与此相比,非A型则包括其他八个族群。51、74和710族群被归为一个簇,而72、86、97和711族群形成了另一个簇。85族群则单独作为一个单一族群簇。非A型族群中,72、85和86是多家族族群。72族群包含7个家族,共28个基因;85族群包含11个家族,共33个基因;86族群包含4个家族,共28个基因;其他族群则是单家族族群,每个族群包含1至3个基因。详细信息可见图1A。
红花中的140个UGT序列被分为15个组,组A、B、C、D、M、O和Q被归为一个簇,组G、H、I、J、K、L和N形成了另一个簇,组E单独形成一个簇。最大组G由30个基因成员组成;而最小组C、J、K和Q每组仅包含2个成员。其他组的成员数如下:组A有14个成员,组B有3个成员,组D有13个成员,组E有26个成员。此外,组H和组I分别有6个成员,组L有15个成员,组N有5个成员,组O有11个成员。详细信息可见图1B。
CYPs和UGTs的保守基序、基因结构与染色体定位
使用MEME软件,我们鉴定出了红花CYP和UGT家族中的20个保守基序。总体来说,这些基序在不同家族或组之间的分布差异显著,而在同一家族或组内则呈现出相似的模式。在CYPs中(图2A,详细信息见图S2和S3),A型和非A型之间有显著的区别,A型中出现了所有20个基序,而非A型CYP中仅出现了6到19号基序。在这些基序中,1、2、4和5号基序在所有CYP族群中都高度保守。具体来说,5号基序主要位于C端,而1、2和4号基序主要位于N端。其中,7个基序(1到7号)包含了功能性已鉴定的结构域。
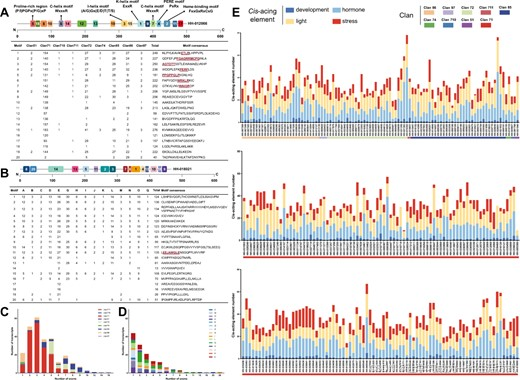
图2 红花CYPs九个族群和UGTs十五个组中的保守基序与外显子分布,以及红花CYPs九个族群启动子区的顺式作用元件数目 (A) 红花CYP蛋白中的保守基序示意图,以HH-012066为例。以及20个保守基序在九个CYP族群中的分布。含有功能性已鉴定结构域的标志性基序被标示。N和C分别代表N端和C端。此外,还展示了九个CYP族群中转录本的外显子分布。 (B) 红花UGT蛋白中的保守基序示意图,以HH-018021为例,以及20个保守基序在十五个UGT组中的分布。 (C) CYP转录本的外显子分布。 (D) UGT转录本的外显子分布。 (E) 预测的顺式作用元件被分为四类:发育、应激、激素和光响应。
基序1包含K-螺旋基序(ExxR)。基序2为血红素结合基序(FxxGxRxCxG)。基序3包含I-螺旋基序((A/G)Gx(E/D)T(T/S))。基序4为PERE基序(PxRx)。基序5包含富含脯氨酸区域((P/I)PGPx(P/G)xP)。基序6和7包含C-螺旋基序(WxxxR)。在UGTs中(图2B,详细信息见图S4和S5),基序的偏好在各组之间有所不同。只有组E包含所有基序,而其他组只包含12-16个基序。在这20个基序中,基序1、2、3、4、5、6、10和11在所有UGT组中高度保守。基序2、3和5主要位于C端,而基序1、4、6、10和11主要位于N端。基序17和18的保守性较差,仅在组E中出现。
CYPs和UGTs的外显子、内含子、编码序列(CDS)和非翻译区(UTRs)的结构分布也被总结,以增强对基因家族结构演化的理解。非A型CYP的CDS-UTR构成与A型CYP相比有显著不同,A型CYP显示了相似的分布(图2C和图S6)。A型CYP包含1至8个外显子,其中59.6%(99个中的166个)含有2至3个外显子。相比之下,非A型CYP的外显子数量波动较大。例如,97家族显示有10个或更多外显子,85家族的外显子数量在2到12个之间,而其余CYP家族一般少于10个外显子。在UGTs中(图2D和图S7),少数基因(140个中的6个)有超过9个外显子,而大多数基因(140个中的96个)包含1至3个外显子。具体来说,组A(140个中的14个)、组D(140个中的13个)和组J(140个中的2个)有1至6个外显子,而组B(140个中的3个)、组C(140个中的2个)、组K(140个中的2个)、组L(140个中的15个)和组O(140个中的11个)有1至4个外显子。
基因在染色体上的定位是通过红花的基因组注释数据进行的(图S8)。在264个CYP基因中,157个基因,140个UGT基因中,111个基因被随机分布在12条染色体上。剩余的基因无法定位到特定的染色体,因此仍然定位在支架上[25]。CYP和UGT基因在染色体上的分布呈现不均匀的模式。在染色体1上观察到CYP和UGT基因的高密度,分别包含56个和47个基因。相反,CYP基因在染色体5和7上的分布最少,每个染色体上只有5个基因。同时,UGT基因在染色体8上的存在最少,只有1个基因。
CYPs和UGTs启动子区顺式作用元件分析
为了更好地分析红花CYP和UGT基因的转录调控机制,我们提取了每个转录本起始密码子(ATG)上游2000个碱基对(bp)的序列,用于顺式作用元件的预测。除了核心元件外,顺式作用元件被分为四类:发育元件、应激相关元件、激素元件和光响应元件。
在32个预测的光响应元件中,频率最高的四个分别是Box 4(90.9%,240个中的264个)、G-box(83.3%)、GT1-motif(78.8%)和TCT-motif(58.7%),这证实了光响应元件是红花CYP基因家族启动子中最为丰富的元件(图2E,更多细节见图S9和S10)。其次是激素响应顺式作用元件,TGACG-motif和CGTCA-motif是最频繁的,每个出现的频率为78.4%。紧随其后的是脱落酸响应元件(ABRE),出现频率为78%,乙烯响应元件(ERE)出现频率为59.5%。在与应激反应相关的顺式作用元件中,ARE(90.9%)、WRE3(54.2%)、LTR(48.9%)和TC-rich重复序列(37.9%)是最丰富的。特别是,ARE是厌氧诱导必需的,WRE3作为伤口响应元件,LTR是低温响应元件,TC-rich重复序列与防御和应激反应相关。在红花CYP中,还鉴定出了几种发育响应顺式作用元件,包括CAT-box、circadian、HD-Zip 1和GCN4_motif,分别与分生组织表达、昼夜节律控制、栅栏细胞分化和胚乳表达相关。这些元件分别出现在90个(34.1%)、58个(22%)、35个(13.3%)和30个(11.4%)CYP中。
UGT启动子序列的分析(见图S11、S12和S13)揭示了光响应元件的显著多样性和丰富性。预测了33个光响应顺式作用元件,其中四个最常见的是G-box(83.6%)、Box 4(78.6%)、GT1-motif(69.3%)和TCT-motif(57.9%)。在红花UGTs中,四个最常见的激素响应顺式作用元件分别是ABRE、TGACG-motif、CGTCA-motif和ERE。这些顺式作用元件分别与脱落酸、MeJA和乙烯响应相关。具体而言,TGACG-motif和CGTCA-motif与MeJA响应相关,它们的出现率分别为82.9%(140个中的116个)、81.4%(140个中的114个),ERE的出现率为58.6%(140个中的82个)。同时,还鉴定出与应激反应相关的顺式作用元件,包括最常出现的厌氧诱导元件(ARE,93.6%)、伤口响应元件(WRE3,57.9%)、低温响应元件(LTR,52.1%)和干旱诱导元件(MBS,40.7%)。还鉴定出了几个发育响应顺式作用元件,包括AAGAA-motif、CAT-box、circadian和GCN4_motif,分别与次生木质部发育、分生组织表达、昼夜节律控制和胚乳表达相关。这些元件在UGTs中的出现率分别为62.1%、30%、19.3%和14.3%。
红花不同部位黄酮类化合物的表型与代谢关联分析
在对红花的根、茎和叶进行横截面切片后,观察到一种明显的红色物质(RS)渗出。随后,在荧光显微镜下观察红花根的横截面,发现RS在内皮层和形成层之间呈现出明显的绿色荧光(图3和图S19)。UPLC结果显示,RS和HSYA的保留时间完全不同,MS结果则表明,RS和HSYA在分子量和质谱方面存在显著差异,表明RS和HSYA是两种完全不同的化合物。此外,代谢分析显示,HSYA在红花根、茎和叶的黄酮类代谢谱中并不存在,这进一步表明HSYA是红花花朵中特异性生物合成的化合物(表S5和表S6)。
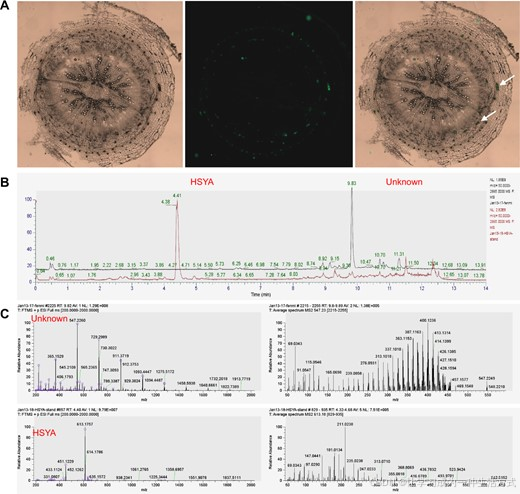
图3 红花根、茎和叶中RS的鉴定 (A) 红花根中RS的分布。从左到右分别为明视野、绿色荧光蛋白荧光和合并照明图。 (B) RS和HSYA的HPLC色谱图。 (C) RS和HSYA的质谱图。
红花在不同发育阶段、光照强度、组织和MeJA处理下的CYP和UGT基因表达谱
许多因素影响基因表达。启动子分析表明,发育、光照、激素和应激是影响CYP和UGT基因表达的主要因素。在我们的初步研究中,我们发现发育阶段、光照强度和MeJA处理显著影响红花中两类黄酮糖苷的水平,此外,不同组织中的黄酮糖苷类型和含量差异显著。顺式作用元件分析进一步证实了CYP和UGT参与红花黄酮糖苷的生物合成。因此,分析这些条件下CYP和UGT的表达模式将有助于在未来的研究中识别功能基因。
我们分析了红花在不同开花阶段、不同光照强度、不同组织和MeJA处理下的转录组数据(图S14和图S15)。根据表达模式,CYP和UGT基因分别被分为不同的簇,每个簇由显示相似表达谱的基因组成。不同光照强度和不同发育阶段下的基因表达谱被分类为六个簇。不同光照强度和不同发育阶段下的基因表达谱被分为六个簇,每个CYP簇包含15–69个基因,UGT簇包含13–32个基因(图S16A和S16B)。用MeJA处理的样本以及来自不同组织的样本也被分析并分为六个簇。每个CYP簇包含17–63个基因(图S16C),每个UGT簇包含11–31个基因(图S17C)。
在红花花朵在不同光照强度处理下的转录组簇分析中,127个CYP基因(来自簇2、4和5)和60个UGT基因(来自簇3、4和6)的表达显示出先增加后下降或逐渐上升的趋势;CYP包括51和97族群的所有成员,86族群的64.29%成员,以及71族群的47.59%成员;此外,UGT包括B、I、L、M和O组的60%以上成员。在花朵1至4阶段的转录组数据簇分析中,我们专注于那些显示整体表达水平上升趋势的基因;在簇2、4和5中共鉴定了116个CYP基因,其中71族群成员最多,占63.79%;在簇2、5和6中共鉴定了59个UGT基因,主要来自E组(18.64%)、G组(27.12%)和L组(18.64%)。考虑到红花的黄酮成分在花朵中丰富,并可能受MeJA诱导,我们重点关注簇4、5和6中的CYP基因(103个基因)和簇3、4和5中的UGT基因(65个基因);在选定的CYP基因中,71族群的成员最多,占57.28%;在选定的UGT基因中,除了B组、C组和Q组外,其他组的成员比例从3.08%到21.54%不等。
红花中的尿苷二磷酸糖苷转移酶的发现
植物二次代谢产物的空间或时间特异性产生通常与一组空间或时间特异性生物合成通路基因相关。先前的研究表明,HSYA和山奈酚的积累在光照强度升高时呈现先增加后下降的趋势。红花中黄酮糖苷的含量在开花的第三到第四天达到最高水平,并且在响应不同开花时间时表现出相应的变化。根据这些黄酮糖苷的积累模式,我们筛选出了显示相似表达趋势的基因。通过对三种条件下筛选出的基因进行维恩图分析,最终识别出26个CYP基因和17个UGT基因(图4A–C)。
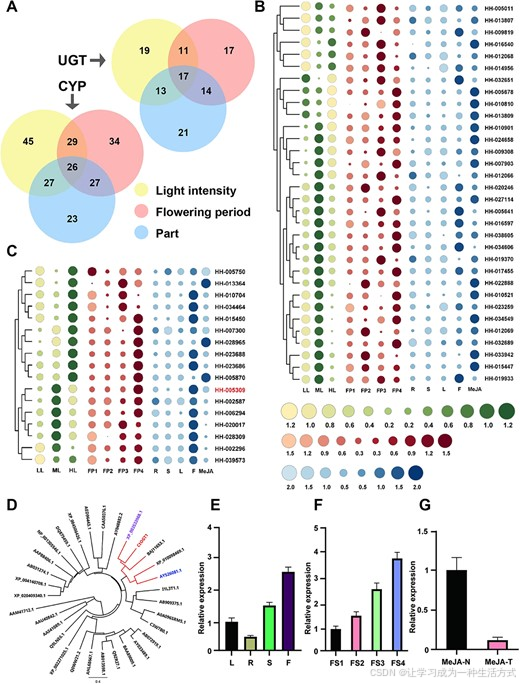
图4 红花中与黄酮类生物合成相关的26个CYP基因和17个UGT基因的表达模式,以及CtOGT1的进化和表达分析 (A) 每个表达分析中CYP和UGT的数量。 (B) 不同开花阶段、不同光照强度处理和MeJA处理下CYP的表达模式。 (C) 不同开花阶段、不同光照强度处理和MeJA处理下UGT的表达模式。 (D) CtOGT1与其他30个OGTs的进化关系。使用最大似然方法构建该树,并通过1000次自助法支持。树根采用AAM41712.1。使用的蛋白质的GenBank ID及其物种名称:AAM41712.1,黄单胞菌;AAU40842.1,地衣芽孢杆菌;AAS41089.1,蜡样芽孢杆菌;Q9LNE6.1,拟南芥;XP_002271025.1,葡萄;Q0WW21.1,拟南芥;AHL68667.1,北方葡萄;AB013598.1,香雪球;Q9ZR27.1,紫苏;BAA89009.1,桥头金鱼草;AY033489.1,茄子;AB072919.1,烟草;C3W7B0.1,印度稻;A0A096SRM5.1,玉米;AB909375.1,荞麦;I1L3T1.1,大豆;AY526081.1,甜菜;XP_010098469.1,桑树;ABJ11653.1,绿脓假单胞菌;XP_003533968.1,大豆;AY048882.1,柚子;CAA50376.1,桥头金鱼草;AED96443.1,拟南芥;XP_004506426.1,鹰嘴豆;DQ875459.1,紫花苜蓿;XP_001305546.1,阴道毛滴虫;AAP88406.1,洋葱;AB031274.1,黄芩;XP_004140708.1,黄瓜;XP_020409340.1,桃子。 (E) 不同组织中的表达。L,叶;R,根;S,茎;F,花。 (F) 花发育的四个阶段中的表达。FP1,花发育的第一阶段;FP2,花发育的第二阶段;FP3,花发育的第三阶段;FP4,花发育的第四阶段。 (G) MeJA处理下的表达。MeJA-N,无MeJA处理;MeJA-T,MeJA处理。
红花中鉴定的CYP和UGT基因与已知功能的同源基因进行了系统发育分析,如图S18所示。结果表明,26个CYP基因与其47个同源基因的相似性较低(图S18A)。在包含30个同源基因的UGTs系统发育树中,HH-005309与BAJ11653.1和XP_003533968形成一个分支(图S18B)。此外,HH-005309与来自大豆的XP_003533968关系更为密切,XP_003533968是7-O糖苷转移酶家族的成员(图4D)。因此,HH-005309被命名为CtOGT1。表达分析表明,CtOGT1表现为在花朵中主要的表达,且在花发育的晚期阶段表达最高。我们还观察到,MeJA处理能够抑制CtOGT1的表达(图4E–G)。
CtOGT1的功能验证
植物内源基因功能验证的广泛研究方法之一是利用农杆菌渗透法对基因进行功能验证[26]。在本研究中,我们首先使用这一系统验证了CtOGT1的功能。我们将桔梗黄酮(图5A)和芹菜素(图5B)作为底物,观察到当同时加入底物和CtOGT1时,黄酮糖苷的转化率较高,而仅加入底物时转化率较低,这表明CtOGT1具有糖苷转移酶活性。然而,即使没有添加CtOGT1,我们也在烟草的叶片中检测到了黄酮糖苷,提示植物叶片中存在内源性的GT。
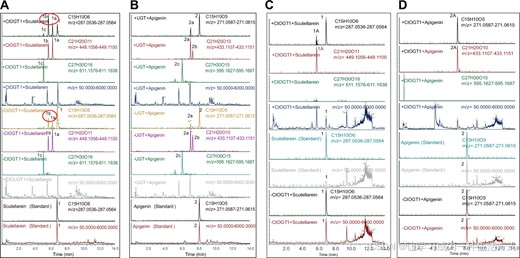
图5 通过烟草农杆菌渗透法和原核表达验证CtOGT1功能,使用桔梗黄酮和芹菜素作为底物 (A) 使用桔梗黄酮作为底物在烟草中验证CtOGT1功能。-CtOGT1表示未注射含有该基因的农杆菌,+CtOGT1表示注射了含有该基因的农杆菌。+scutellarin表示注射了底物。我们以桔梗黄酮的标准作为对照。根据分子量和MS2分析,我们可以知道1a是一个糖苷附加到桔梗黄酮,1b也是一个糖苷附加到桔梗黄酮,1c是两个糖苷附加到桔梗黄酮。图中的红色圆圈显示了当转化了CtOGT1后,产物的转化率得到了提高。 (B) 使用芹菜素作为底物在烟草中验证CtOGT1功能。+Apigenin表示注射了底物。我们以芹菜素的标准作为对照。根据分子量和MS2分析,我们可以知道1a是一个糖苷附加到芹菜素,1b是一个糖苷附加到芹菜素,1c是两个糖苷附加到芹菜素。图中的红色圆圈显示了当加入CtOGT1时,产物的转化率提高了。 (C) 使用桔梗黄酮作为底物通过原核表达验证CtOGT1功能。+CtOGT1 +scutellarin表示同时存在CtOGT1和桔梗黄酮。-CtOGT1 +scutellarin表示没有CtOGT1,但有桔梗黄酮。我们还检测了桔梗黄酮的标准。根据分子量和MS2分析,我们可以看到1是桔梗黄酮,1A是桔梗黄酮-7O糖苷,并且只生成了一个产物。 (D) 使用芹菜素作为底物通过原核表达验证CtOGT1功能。+CtOGT1 +apigenin表示同时存在CtOGT1基因和芹菜素。-CtOGT1 +apigenin表示没有CtOGT1,但有芹菜素。根据分子量和MS2分析,我们可以知道1是芹菜素,1A是芹菜素-7O糖苷,并且只生成了一个产物。
因此,我们使用了原核表达系统,这是一种广泛用于鉴定糖苷转移酶(GT)的系统[27, 28]。最初,我们测试了诱导条件并证明了CtOGT1可以成功诱导并在上清液中检测到。我们使用粗酶液评估了CtOGT1对桔梗黄酮和芹菜素的活性,结果表明仅生成了一个新的产物。为了确定糖苷化的位置,我们将这个产物与桔梗黄酮糖苷和芹菜素糖苷的标准进行了比较。分析结果显示,糖苷化发生在黄酮分子的第7位羟基上,因为新的产物与桔梗黄酮7-O糖苷和芹菜素7-O糖苷的保留时间一致。我们的结果表明,该酶只能在黄酮类化合物上添加一个糖苷,特别是桔梗黄酮和芹菜素(图5C,D)。
CtOGT1的动力学参数分析
我们首先纯化了CtOGT1,并通过Western blot分析评估了纯化效果(图6A)。随后,使用纯化的蛋白质评估了CtOGT1在不同底物上的动力学参数,包括桔梗黄酮(图6B)、芹菜素(图6C)、山奈酚(图6D)和山奈酚(图6E)。结果表明,Km值分别为桔梗黄酮48.51、芹菜素32.93、山奈酚74.16、山奈酚59.67。对应的Vmax值分别为19.82、6.892、23.08和12.48 μM·min−1 μg−1。
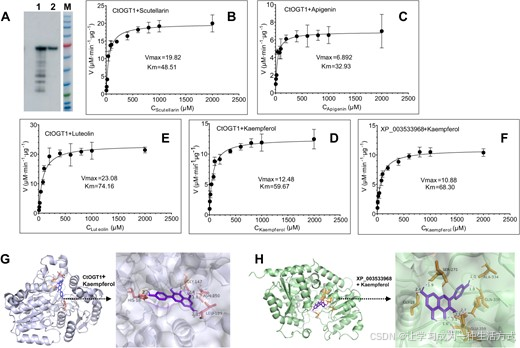
图6 CtOGT1的动力学参数分析和CtOGT1与山奈酚的分子对接 (A) CtOGT1的Western blot分析,1表示转化了CtOGT1的大肠杆菌总蛋白的Western blot,2表示纯化的CtOGT1蛋白的Western blot。 (B) 使用桔梗黄酮作为底物对CtOGT1进行动力学参数分析。 (C) 使用芹菜素作为底物对CtOGT1进行动力学参数分析。 (D) 使用山奈酚作为底物对CtOGT1进行动力学参数分析。 (E) 使用山奈酚作为底物对CtOGT1进行动力学参数分析。 (F) 使用山奈酚作为底物对XP_003533968进行动力学参数分析。 (G) CtOGT1与山奈酚的分子对接。 (H) XP_003533968与山奈酚的分子对接。 图中展示了整体和部分图像,并示出了氢键的位置和距离。红色圆圈的位置是黄酮类化合物的7-OH位置。
此外,我们从大豆中克隆了XP_003533968,该基因也已知对黄酮类化合物的7-OH位置具有活性[29],并使用山奈酚作为底物,比较了XP_003533968和CtOGT1的酶活性。结果显示,XP_003533968对山奈酚的Km值为68.30,Vmax为10.88 μM·min−1 μg−1(图6F)。这表明CtOGT1对山奈酚具有更高的亲和力和敏感性。为了进一步探究这一发现,我们使用AlphaFold预测了这两种蛋白质的结构,并使用AutoDock进行了分子对接模拟(图6G,H)。结果表明,底物在两种酶中都位于中心位置。CtOGT1与山奈酚第7位的羟基形成了一个氢键,而XP_003533968蛋白在相同位置形成了两个氢键。这表明,CtOGT1在该位置为糖苷结合提供了更多的空间。此外,CtOGT1的氢键距离为2.1 Å,比XP_003533968的2.4 Å更近,暗示CtOGT1的Km值较低。CtOGT1与桔梗黄酮、芹菜素和山奈酚的分子对接结果见图S20。
讨论
红花是一种重要的经济作物,其花朵富含黄酮类糖苷。然而,特有的红花查尔酮糖苷的生物合成机制仍不明确,目前提出的生物合成途径存在争议。例如,本研究假设UGT催化中间化合物转化为HSYA,而吴等人[30]则提出HSYA是由CYP和UGT共同催化中间化合物形成的。在这两种假设中,CYP和UGT是催化红花黄酮糖苷形成的关键基因,并且影响糖苷的多样性。因此,本研究结合全基因组和多组学分析,并通过不同组织中的表型筛选,识别了参与红花黄酮糖苷生物合成的关键基因。
CYPs涉及多种功能,包括膜甾醇、植物激素、信号分子、紫外线保护剂和挥发性有机化合物的形成,这些物质有助于生物与非生物的相互作用,强调了其对植物生理过程的重大影响[31]。在红花基因组中共鉴定出264个CYP基因,分为44个家族,并归类为9个族群;然而,族群727中没有基因。CYP基因的数量和分类在不同植物物种间差异较大。例如,研究发现从大花石斛中鉴定出226个CYP基因,分为8个族群[32],而从金线莲中鉴定出的257个CYP基因分为9个族群[32],Ankur等人从阿卡普库河木材基因组中鉴定出136个CYP基因并将其分配到8个族群和38个家族[33]。与大多数被子植物类似,四个多家族的族群(71、72、85和86)是最大的,占红花总CYP基因的96.6%。植物CYPs分为两类:A型,包括71族群,和非A型,包括所有其他族群[34]。红花的CYP基因,按照A型和非A型分类,显示出基因结构和保守基序的明显差异[35]。与A型CYPs相比,非A型CYPs在这两个方面的变异性较大。然而,四个基序在两类CYP中都高度保守:K-螺旋基序(基序1)、血红素结合基序(基序2)、I-螺旋基序(基序3)、PERE基序(基序4)和富含脯氨酸区域(基序5)。这些基序对红花CYP酶的催化功能至关重要。血红素结合基序使CYP蛋白能够高效催化黄酮类化合物的氢氧化反应。K-螺旋基序确保了黄酮在酶内的准确定位,而I-螺旋基序则促进了氧的激活,从而使氢氧化过程得以进行36。此外,大多数非A型CYPs在C端缺少四个特定基序(基序5、11、13、15)以及在N端缺少一个基序(基序16)。这五个基序在A型CYPs中普遍存在,并被认为在A型CYPs中发挥重要作用。总之,红花CYPs的结构多样性可能是其多种生理活动发展的原因,这些活动使得红花能够生物合成广泛的活性黄酮类化合物。CYP基因家族在生物体内具有广泛和多样的功能,将CYP基因归类到特定家族有助于理解它们在植物代谢网络中的作用。这些族群中的97、74、710和51族群与植物的主要代谢相关[38],97、72、711、85、74、51和71族群的基因参与植物对环境胁迫的反应[39]。86、97、72和71族群的基因可以影响植物对激素的反应、合成或代谢[40]。51、71、85和86族群的成员参与二次代谢,可以影响黄酮、萜类和生物碱的合成[41]。例如,CYP76和CYP706已知催化萜类的生物合成,而CYP75和CYP93家族通常催化黄酮的生物合成[42]。鉴于红花特有的黄酮糖苷生物合成,CYP基因家族的详细和全面的数据集对于识别和筛选功能基因至关重要[36]。
GTs催化糖苷分子从供体分子转移到受体分子的过程,其中UGT是这一组中最为广泛的基因家族[43]。UGTs发挥多种作用,包括调节生长和发育、防御病原和非生物胁迫以及适应不同环境[44]。基于系统发育关系,UGTs目前可分为18个组。阿拉伯芥中识别出了A至N编号的14个组[45],而在苹果[46]、玉米[47]和橙子[48]中发现了另外四个编号为O至R的组。不同物种中UGTs的分组和数量存在显著差异。例如,在红毛榉基因组中鉴定出152个UGT基因,并分为13个组[49],而在吉萨棉基因组中鉴定出的157个UGT基因被分为15个组[44]。在本研究中,从红花基因组中鉴定出了140个UGT基因,并将其分为15个组。这个分类包括之前在阿拉伯芥中发现的13个组和两个新发现的组。值得注意的是,未发现任何基因被归类到F、P和R组。如在阿拉伯芥、油菜、玉米和大豆中报道,与多酚类和黄酮类合成相关的UGT蛋白通常富集在A、E、F、L和M亚组中。与萜类代谢相关的蛋白富集在D和M亚组中,而与植物激素相关的蛋白则富集在H、L、K和O亚组中。此外,G至N组主要涉及植物二次代谢、碳水化合物代谢和脂质代谢的酶[50]。保守基序的数量和分布可以揭示基因家族成员之间的结构相似性和差异,是红花UGT基因家族系统发育分类的重要基础,分析了20个保守基序,发现8个基序(基序1、2、3、4、5、6、10和11)在所有组中都有,表明它们在UGT基因家族的组成中可能具有重要作用。相反,其余14个基序可能在定义亚组中起着重要作用。UGT蛋白中的保守基序对保持酶的整体结构稳定性及其与黄酮底物或糖供体的结合至关重要。例如,D/EX7E基序(基序11,D/ExxxxxxE)通过与底物和糖供体的相互作用促进糖苷基团的转移[51]。外显子/内含子结构的变化可能导致基因或蛋白功能的变化。在本研究中,观察到同一组中的大多数UGT基因具有相似的外显子/内含子结构和数量,这与烟草[50]、紫花苜蓿L.[25]和小麦[52]等其他植物一致。像大多数植物一样,红花中的黄酮主要以糖苷的形式存在。这些黄酮糖苷的多样性受到糖苷化位置(如羟基或羰基)和糖供体类型(如葡萄糖、半乳糖或鼠李糖)的强烈影响。这些因素对于分类UGT组并理解红花UGT基因家族的分类至关重要,有助于节省时间和资源进行目标基因筛选。对UGT家族的详细了解有助于识别红花中参与黄酮糖苷生物合成的特定酶[53]。
我们之前的研究发现,在MeJA处理后,查尔酮糖苷的含量显著增加;黄酮糖苷在开花的第一到第三或第四天上升;它们的含量随着光照强度的变化而波动。在CYP和UGT的启动子分析中,鉴定出了大量与光照、生长、激素和应激相关的顺式作用元件,表明CYP和UGT基因都对这些因素产生反应,进一步证实了CYP和UGT在红花黄酮糖苷生物合成中的关键作用。根据不同条件下CYP和UGT的基因表达模式,这些基因被分为不同的组。同时,根、茎和叶的表型和代谢分析证实了红花中黄酮糖苷的分布存在明显的组织特异性。此外,查尔酮糖苷仅在红花花朵中合成。因此,本研究结合了基因表达分析和黄酮积累模式进行候选基因筛选。然而,进化分析表明,候选的红花CYP基因未与之前鉴定的CYP基因聚类,表明这些候选CYP可能不具备功能。在UGT的进化分析中,发现一个UGT(HH-005309,命名为CtOGT1)与两个已鉴定的UGT基因紧密聚类,表明该基因很可能属于7-O-GT家族。
大多数糖苷转移酶具有广泛的底物专一性,使它们能够从多种底物中生成多样的糖苷化产物。例如,来自雷公藤的O-GT,TwUGT2,专门作用于黄酮的3-OH和7-OH基团[54];有报告显示,从甘草中筛选出了43个OGT候选基因,其中11个已被鉴定,包括异黄酮7-OGTs、黄酮醇3-OGTs和广谱OGTs[55]。然而,我们筛选的基因只能将一个糖苷添加到底物上。我们发现所有三种底物都可以被糖苷化。其他GT可能参与在不同位置添加糖苷。因此,彻底研究O-GTs对不同底物和位置的专一性对于全面了解其生物学活性至关重要。我们的结果表明,原核表达系统是研究GT的有效方法,无论是使用粗酶液还是纯化酶。对于大规模筛选,可以直接使用粗酶液进行活性分析。此外,我们对其他三个底物进行了分子对接:桔梗黄酮、芹菜素和山奈酚(图S2),并在HIS16、LEU-189、ASN-150和GLY147处识别了潜在的活性位点。未来的研究可能会探索突变以优化酶活性。
本实验的另一个有趣发现是,尽管先前的研究表明MeJA促进黄酮的生物合成,特别是如HSYA等C-糖苷类成分,但MeJA抑制了UGT基因的表达[56]。可能在红花中C-糖苷和O-糖苷的调控模式存在差异。郭等人先前的研究表明,三种UGT与O-糖苷密切相关,尽管它们的功能尚未得到确认。此外,这三种UGT的表达模式显示,所有这些基因在MeJA诱导下都受到抑制[57]。因此,进一步研究O-糖苷和C-糖苷生物合成的调控模式对于加深我们对它们在红花中的差异的理解至关重要。对这些机制的深入研究可能为揭示促进黄酮生物合成的复杂途径和优化其生物活性提供重要线索。
CYP和UGT酶在红花黄酮糖苷的生物合成、分布和多样性中发挥着关键作用。本研究采用结合全基因组和多组学分析的综合方法,识别了负责红花黄酮糖苷生物合成的关键基因。研究包括对红花基因组中264个CYP和140个UGT基因家族成员的基因家族分析,探讨了它们的系统发育关系、保守基序、基因结构、顺式作用元件和染色体定位。该分析提供了详尽的基因家族数据。根据红花CYP和UGT基因在不同发育阶段、光照条件、组织和MeJA处理下的表达模式,筛选出了候选基因。随后,通过进化分析与已报道基因的比对,确定了一个活性较高的UGT基因。最终,通过功能验证和酶活性研究,成功筛选出一个功能基因。总之,本研究通过结合全基因组和多组学方法,成功筛选出一个功能基因,并为进一步研究红花黄酮的生物合成和途径分析奠定了基础,为后续的分析和功能解释提供了宝贵的研究思路和数据基础。
材料与方法
红花基因组中CYPs和UGTs的鉴定
红花基因组数据来源于我们之前的研究[12]。简要地,为了鉴定红花中的CYP和UGT基因家族,我们查阅了阿拉伯芥信息资源(TAIR - Arabidopsis),获取了阿拉伯芥中CYP和UGT的序列。此外,CYP(PF00067)和UGT(PF00201)的保守结构域的隐藏马尔可夫模型(HMM)来自PFAM 35.0(Pfam is now hosted by InterPro)。这些模型作为查询在HMMER 3.2.1中用于对红花蛋白序列进行hmmsearch,应用e值阈值为0.1(HMMER)。使用来自拟南芥的氨基酸序列作为查询序列,对红花蛋白数据库进行了本地BLASTP搜索,搜索时的e值阈值为1e-5。通过NCBI保守结构域数据库工具进一步验证了所鉴定的CYP序列(蛋白质长度范围为300到650个氨基酸)及所有UGT序列。
通过TBtools软件[58]计算了CYPs和UGTs的物理化学参数,如氨基酸数目、分子量、理论等电点、不稳定性指数、脂肪指数和水合性总平均值。随后,通过在线平台Plant-mPLoc(Plant-mPLoc server)对这些序列进行了亚细胞定位预测。
系统发育分析、保守基序、基因结构和染色体定位
CYP和UGT的蛋白质序列分别使用MUSCLE算法(集成于MEGA软件中,Home)进行比对。使用TBtools软件[59]的trimAl功能自动修剪了低相似度的区域。然后,通过最大似然(ML)方法从修剪后的比对中生成系统发育树,使用5000次自助法计算了bootstrap值。最终,通过在线平台EVOLVIEW(https://evolgenius.info/evolview-v2/#login)[60]对构建的系统发育树进行了注释并可视化展示。
使用MEME(MEME - Submission form),一款专门用于基序鉴定的在线工具[61],检测了CYP和UGT中的保守基序。参数设置如下:要鉴定的基序数量设置为20,最小基序宽度为6,最大基序宽度为50。通过使用红花基因组的GFF文件确定CYP和UGT的基因结构和染色体位置。通过TBtools辅助可视化和修改保守基序、基因结构和染色体位置。
启动子中的顺式作用元件分析
为了分析顺式作用元件,我们从红花基因组中提取了CYP和UGT基因的起始密码子(ATG)上游2000个碱基对(bp)的序列。这些启动子序列通过PlantCARE数据库(PlantCARE, a database of plant promoters and their cis-acting regulatory elements)进行了分析,用于预测潜在的顺式作用元件。我们重点关注与发育、应激、激素和光响应相关的元素,并保留这些元素以便进一步可视化和分析。
红花的黄酮成分分析
红花植物在中国成都的成都中医药大学温江校区的药用植物园内栽培。红花的根、茎和叶经过横截面切割后,允许液体RS渗出30秒。随后,轻轻吸取这些RS并溶解在甲醇中。样品和HSYA标准品使用超高效液相色谱(UPLC)和质谱(MS)进行分析。在代谢分析中,红花组织先在真空下冷冻干燥,然后使用研磨机(MM 400,Retsch)以30 Hz的频率研磨1.5分钟,得到粉末。接着,称取50 mg粉末样品,加入1200 μL预冷(−20°C)的70%甲醇水溶液,按照每50 mg样品添加1200 μL溶液的比例。混合液每30分钟震荡30秒,共重复震荡六次。经过12000 rpm离心3分钟后,小心吸取上清液,并通过0.22 μm微孔膜过滤。过滤后的溶液被储存在注射瓶中,供随后的UPLC-MS/MS分析使用。红花的根、茎和叶样品在红花抽苞后收集,每个样品来自三株不同的植物。对于红花根、茎和叶的代谢分析,使用一个重复样本,实验步骤遵循我们之前的研究[62]。红花花朵的代谢组学数据来源于我们之前的分析,每个样品来自10朵不同的花,且进行了三次重复实验[56]。
红花的转录组分析
在开花期间,选择了四个发育阶段。第一阶段对应花后第一天(FP1),第二阶段对应花后第二天(FP2),第三阶段对应花后第三天(FP3),第四阶段对应花后第四天(FP4)。同时,收集了根、茎、叶和花样品。MeJA处理根据我们之前的研究进行[56]。简而言之,将100 μM的MeJA溶液(Sigma-Aldrich,瑞士)喷洒在三天后的花朵上,然后将花朵放入透明塑料袋中。经过6小时的处理后,收集花样品。每个花样品的实验进行了三次重复,并收集了三个样本。根、茎和叶的样本作为一个重复样本,并立即用液氮冷冻以保存用于RNA提取和后续的表达分析。此外,MeJA和不同光照强度(低光强度LL;中光强度ML;高光强度HL)处理的红花转录组数据来自我们之前的研究[56, 63]。在本研究中,我们从这些转录组数据集中收集了红花CYP和UGT基因的表达数据。这些数据随后转换为FPKM(每百万条映射片段的外显子每千碱基的片段数)值以进行进一步分析。根据表达水平对基因进行了分组,并使用Chiplot工具(ChiPlot)生成了表达热图。根据这些基因的表达模式,使用在线软件(Home)对它们进行分类。对所选簇中的基因进行Venn分析(jvenn)[64]。最后,对在三种条件下共同选择的基因进行进化分析,以识别候选功能基因。
候选基因的表达模式分析和分离
通过实时PCR确定四个器官、不同发育阶段的花朵以及MeJA处理样品的基因表达。样品采集后迅速用液氮冷冻,并使用TRIzol Chaperone Kit(Tiandz,北京,中国)提取总RNA。通过逆转录从分离的RNA合成cDNA。然后对这些cDNA样品进行实时PCR,使用表S7中列出的基因特异性引物。按照既定的协议进行操作,并通过琼脂糖凝胶电泳确认PCR反应的特异性。所有实验均进行三次重复。
转基因载体的构建与农杆菌渗透法的应用
CtOGT1(HH-005309)的CDS序列从基因组中提取,并使用表S7中列出的引物进行特异性扩增。随后,CtOGT1被克隆入pMD19-T载体(Takara,北京,中国)并进行测序以确认其准确性。CtOGT1基因首先通过BP反应使用Gateway BP Clonase II酶混合物(Thermo Fisher,MA,美国)克隆到pDONR207载体中。接下来,CtOGT1基因通过LR反应引入pEAQ11载体,使用表S7中列出的引物。 N. benthamiana的农杆菌渗透过程涉及几个关键步骤。首先,从新鲜菌落或甘油保存库中培养细菌,培养基为10 ml含选择性抗生素的LB培养液。将该培养液以2000 rpm离心15分钟,弃去上清液。然后,将细胞重悬于15 ml农杆菌渗透液中,并以2500 rpm离心15分钟。 去除上清液后,将细胞重悬于含200 μM乙酰杨梅素的10 ml农杆菌渗透液中。在室温下,温和震荡2小时并在黑暗环境中处理。随后,使用光密度(OD600)测量每个培养液,并用相同的农杆菌渗透液将其调整至0.2的浓度。使用1 ml注射器,将0.5–1 ml的混合液渗透到N. benthamiana叶片的背面。通常在4–6天后收集叶片进行成分分析。
N. benthamiana叶片成分的提取与检测
本研究使用的叶片提取方法基于先前的研究[52]。简而言之,称取10 mg冷冻干燥叶片样品,使用微天平精确称量,并放入2 ml带螺旋盖的微量离心管中,加入两颗钨珠。使用组织研磨器在1000 rpm下均质1分钟,随后在20000 g下短暂离心。接下来,向每个样品中加入400 μl 50%甲醇,置于超声波浴中,设置50%的振幅,温度为4°C,处理时间为15分钟,20秒开/40秒停的间歇。每个样品随后转移到新鲜的1.5 ml微量离心管中,并尽量避免携带叶片物料。样品进一步处理后使用滤管过滤,并以12500 g离心30秒。最终,将50 μl滤液转移至含额外50 μl 50%甲醇的自动进样瓶中。 数据采集使用Thermo Scientific™ UltiMate 3000 UHPLC和Q Exactive台式Orbitrap质谱仪进行。使用的色谱柱为Agilent公司的Eclipse Plus C18(150 mm × 3.0 mm,1.8 μm)。流动相由纯水和0.1%甲酸(A)及乙腈(B)组成。洗脱梯度设置如下:从0到25分钟,B相从5%升至95%,流速为0.2 mL/min。在280 nm波长处记录吸光度,并在正离子模式下获取ESI-MS谱图,生成[M + H]+或[M + Na]+离子,使用干气N2(流速800 L/h,温度450°C,喷射压力6 bar,毛细管电压3 kV)。质量扫描范围为m/z 100到800,目标化合物的分子量通过提取离子色谱图(EICs)来确定。
重组蛋白的表达、纯化与WB分析
CtOGT1基因的原核表达依赖于pET32a载体(Thermo Fisher,美国),使用表S1中提供的引物。将重组载体pET32a-CtOGT1转化入BL21(DE3)菌株。BL21细胞在含50 μg/ml氨苄青霉素的LB培养基中培养,直到OD600达到0.5。然后,使用120 μM IPTG诱导细胞,在16°C下震荡培养16小时。培养后,收集细胞并重悬于30 ml裂解缓冲液(25 mM HEPES,pH 8,500 mM NaCl,5 mM咪唑)中进行冰上超声破碎。超声后,混合物经过离心去除上清液。所得的细胞沉淀用于酶活性检测。
通过纯化酶来评估其动力学参数。为此,超natant经过0.45-μM滤膜过滤,然后加入Ni-NTA树脂(Qiagen,德国),比例为1.5 ml树脂/每升细胞培养液,随后在4°C下孵育1小时。混合液被应用于重力流动柱中,弃去流出液,使用洗涤缓冲液(25 mM HEPES,pH 8,100 mM NaCl,20 mM咪唑)洗柱。通过洗脱缓冲液(25 mM HEPES,pH 8,100 mM NaCl,250 mM咪唑)洗脱标记蛋白。最终,使用Bradford法检测浓度。通过30 kDa Amicon Ultra旋转滤器浓缩CtOGT1,并加入10% v/v甘油。冷冻后,将蛋白储存于−80°C。按照先前的报告[65]对CtOGT1进行Western blot分析。
原核表达反应产物的检测
首先,使用原核表达的粗酶液测试了CtOGT1的糖基转移活性。通过与相应的糖苷标准品进行比较,使用UHPLC分析反应产物。反应条件包括使用1 mM的受体底物、2 mM的UDP-葡萄糖、10 μM纯化的重组蛋白以及50 mM Na2HPO4-NaH2PO4缓冲液(pH 8)组成的100 μl反应体系。反应在25°C下进行5分钟。反应结束后,迅速混合并均质化200 μl预冷的甲醇,随后以12000 rpm离心15分钟,取上清液并加载到注射瓶中。UHPLC的实验细节如前所述,梯度洗脱过程从0到40分钟B相由5%增至95%,接着在0到25分钟内以0.2 ml/min的流速进行。随后,我们使用Thermo Scientific™ UltiMate 3000 UHPLC和Q Exactive台式Orbitrap质谱仪(Thermo Fisher,美国)对产物进行表征。将3 μl样品注入离子源,喷雾电压为3.2 kV,离子源温度为350°C,包层气流速设为35 arb,辅助气流速为10 arb,离子传输管温度保持在320°C。对于正离子检测,我们采用全MS/DD-MS2扫描模式,第一级分辨率为35,000,第二级分辨率为17,500,扫描范围为m/z 100到1500,采用20、40和60 eV的碰撞能量梯度。
酶动力学参数的测定
为了测定红花糖苷转移酶重组蛋白催化山奈酚糖苷化的动力学参数,我们建立了一个反应体系,使用50 mM Na2HPO4-NaH2PO4(pH 8)缓冲液,加入过量的UDP-葡萄糖,并设定重组蛋白浓度为1 μM。受体底物山奈酚-7-O-糖苷的浓度从5 μM至1200 μM不等。反应在25°C下进行5分钟,反应完成后迅速通过加入200 μl预冷甲醇并在12000 rpm下离心15分钟进行终止。收集上清液并加载到注射瓶中,通过UPLC定量产物含量,检测条件如前所述。实验重复三次。此外,为了测定红花糖苷转移酶重组蛋白对底物的动力学参数,使用类似的反应体系,选择山奈酚-7-O-糖苷作为中间产物来测定受体底物。反应在不同浓度的底物(5 μM至1500 μM)和过量UDP-葡萄糖的条件下进行,使用相同的缓冲液和蛋白浓度,并在25°C下进行5分钟。反应通过甲醇迅速终止,并使用UHPLC定量产物含量,检测条件与前述相同。实验重复三次。这些分析程序对于准确测定红花糖苷转移酶重组蛋白对底物的动力学参数,并为进一步实验选择最佳反应条件是必需的。
分子对接
使用AlphaFold(AlphaFold Protein Structure Database)构建CtOGT1蛋白的三维结构,而配体的三维结构则从PubChem(PubChem)获取。使用AutoDock Tools [66]对CtOGT1与黄酮类化合物(桔梗黄酮、芹菜素、山奈酚和山奈酚)的分子对接进行分析。通过PyMOL [67]对建模的分子对接结果进行可视化展示。