梨泛转录组-文献精读145
Pan-transcriptome analysis provides insights into resistance and fruit quality breeding of pear (Pyrus pyrifolia)
全转录组分析为梨(Pyrus pyrifolia)的抗性与果实质量育种提供了新的见解
比较转录组学方法推断基因共表达网络及其在玉米和水稻叶片转录组中的应用 TO-GCN时序分析-文献精读-8-CSDN博客
CYP76AKs的功能分化塑造了鼠尾草属中松香烷型二萜化合物的化学多样性-比较转录组-文献精读12_functional divergence of cyp76aks shapes the chemo-CSDN博客
比较转录组分析揭示了116种山茶属(Camellia)植物的深层系统发育和次生代谢物演化-文献精读分享1_泛转录组-CSDN博客
多组学研究揭示梨果实代谢中DNA甲基化的调控机制-文献精读84_multi-omics provide insights into the regulation o-CSDN博客

亮点
首次构建了沙梨全转录组,使用了来自不同组织的506个Pyrus pyrifolia样本,共包含156,744个转录本,其中新转录本在防御反应中表现出显著富集。 揭示了基于表达存在/缺失变异(ePAVs)的表型之间的内在关系以及育种过程中对抗病性的选择。 共表达网络分析表明,梨的石细胞形成、花青素合成和抗病性受到多个模块和基因的共同调控
摘要
Pyrus pyrifolia,常被称为沙梨,是温带地区重要的经济水果树种,拥有丰富的遗传资源,有助于梨的果实品质改良。然而,关于梨等水果物种在抗性与果实质量性状之间关系的研究仍然有限。全转录组能够有效捕捉编码区域的遗传信息,并反映个体之间基因表达的变化。在本研究中,我们基于来自沙梨不同组织的506个样本构建了全转录组,并探讨了表型之间的内在关系以及基于表达存在/缺失变异(ePAVs)在育种过程中对抗病性的选择。研究中的全转录组包含156,744个转录本,其中新转录本在防御反应中表现出显著的富集。值得注意的是,抗病基因在梨的地方品种中高度表达,但在该多年生树种的改良过程中已被选择去除。我们发现,遗传多样的地方品种可分为两大亚群,并推测它们经历了不同的扩散过程。通过共表达网络分析,我们确认了梨中石细胞形成、果实花青素合成和抗逆能力之间的相互关系。它们共同受到多个模块的调控,且调控基因的表达与这三种过程具有显著的相关性。此外,我们还发现了可能影响糖分含量的候选基因HKL1,并且该基因在参考基因组中缺失。本研究为复杂果实性状之间的关联提供了新的见解,同时也为梨的抗病性和果实质量育种提供了数据库资源。
1. 引言
梨(Pyrus)属于蔷薇科杏亚科的雄果族,是温带地区最受欢迎的水果作物之一(Wu et al. 2018)。Pyrus属至少包含22个物种,超过5000个种质资源,但仅有五个物种被广泛栽培(Wu et al. 2013)。沙梨(Pyrus pyrifolia)原产于中国西南部,现已广泛分布于韩国、日本和中国长江以南的亚热带季风气候地区(Song et al. 2014)。这些地区复杂的气候和地理条件造就了众多具有独特特征的地方品种(Jiang et al. 2009)。至今,这些高度多样的地方品种和栽培沙梨得到了很好的保护,并在全球范围内栽培。
不同个体之间的显著表型差异在物种中普遍存在。随着下一代测序技术的发展,越来越多的测序报告提供了大量的全基因组信息,可以揭示物种内个体基因组的差异。目前,国家生物技术信息中心(NCBI)参考序列(RefSeq)数据库中收录了来自1350个不同植物物种的3061个基因组。如此众多的参考基因组的发布推动了植物功能基因组学和群体遗传学的研究(Sun et al. 2022)。随着使用SNP(Su et al. 2019;Zhang et al. 2021)、InDels(Liu et al. 2019;Ou et al. 2020)、CNVs(Prunier et al. 2019)和PAVs(Ou et al. 2018;Lee et al. 2022)进行的比较基因组学研究的深入,我们逐渐意识到,单一的参考基因组不足以捕捉物种内的多样性。
全基因组概念首次由Tettelin等人(2005)提出。全基因组整合了物种的遗传信息,便于研究遗传差异,但这些信息并不总是能够反映个体间基因表达的差异。全转录组这一术语最早由Hirsch等人(2014)在玉米中提出,他们发现仅有16.4%的代表性转录本在所有样本中都有表达,并且大部分表达差异未在参考个体中得到体现。与全基因组(Golicz et al. 2016)类似,全转录组是一个物种在基因组编码区域的表达总和,通常基于RNA-seq数据。与单一转录组相比,全转录组能够反映群体中基因的表达特征。通过全转录组分析识别的ePAVs不仅能够反映基因组结构变异,还能反映遗传调控元件的变异(Jin et al. 2016)。近年来,已经为几种植物构建了全转录组,如大麦(Ma et al. 2019)、番茄(Liu et al. 2020)和茶树(Kong et al. 2022;Wu et al. 2022)。这些研究为树木植物全转录组的构建奠定了基础。
多年生果树比许多其他物种具有更高的杂合度和遗传多样性(Khan和Korban 2022)。然而,目前还没有反映任何果树作物中基因表达多样性的全转录组。沙梨具有丰富的表型多样性,包括石细胞含量、果皮颜色、可溶性固形物含量、果形和果香等特征。为了促进分子育种和基因鉴定,研究人员对312个沙梨品种的8个果实品质性状和3个果实物候性状进行了全基因组关联分析(GWAS),并鉴定了与石细胞形成相关的新基因PbrSTONE(Zhang et al. 2021)。最近,一项系统的遗传学研究揭示了梨果实石细胞木质纤维素形成的调控机制,并鉴定了能够调节多个参与石细胞形成的靶基因的PbrNSC基因(Wang et al. 2021)。在这两项研究中,沙梨个体DNA序列数据对齐率较低的一个原因是仅有单一参考基因组,这导致丢失了个体特异性序列信息,从而降低了性状关联效率。构建一个能够代表梨的基因组信息多样性的全转录组有助于解决这一问题,并捕捉基因表达的多样性,从而为提高梨的果实品质提供基础。
抗性与品质性状之间可能存在一定联系(Islam et al. 2003;Moing et al. 2003),但抗病品种通常表现出不利的品质特征(Stoeckli et al. 2011)。因此,平衡抗病性和作物品质是育种中的一大挑战。了解不同种质的独特遗传信息有助于保护物种的遗传多样性并支持抗病性育种。梨的改良起步较晚,育种中的抗病性研究也有限。研究表明,当梨的防御机制被激活时,病程相关基因、过氧化物酶基因(POD,木质素合成的关键酶)和几丁质酶基因(CHI,花青素合成的关键酶)的表达水平显著增加(Sun et al. 2018;Yan et al. 2018)。然而,梨的抗病性与石细胞形成或果色之间是否存在关联仍不明确。
在本研究中,我们基于来自506个不同组织的RNA-seq数据集构建了沙梨的全转录组,以克服单一参考基因组的局限性。通过识别ePAVs,我们表征了群体中基因的表达模式,从而为梨的基因挖掘提供了数据库。此外,我们通过整合转录组学分析,鉴定了梨的抗病性与果实品质性状之间基因表达模块的关联。进一步地,我们构建了与糖合成相关基因的共表达网络,为功能基因的探索奠定了基础。我们的结果为不同性状之间复杂的基因表达模式和关联提供了新见解,有助于解决多年生果树育种中抗病性与果实品质改良之间的矛盾。
2. 材料与方法
2.1. RNA-seq 数据收集
从NCBI下载了481个沙梨(P. pyrifolia)组织的RNA-seq数据集,包括果肉、果皮、叶子、花蕾和花粉。此外,本研究还生成了来自40个额外品种的果肉组织的转录组数据(附录A)。我们在这些品种的果实成熟时采收,并将其冻存于液氮中。使用TRIzol试剂提取总RNA,并根据Illumina提供的说明构建配对末端RNA-seq文库,并使用NovaSeq 6000平台进行测序,获得150 bp的配对末端读取数据。
2.2. 全转录组构建
使用Trimmomatic(v0.39)(Bolger et al. 2014)对原始RNA-seq数据进行修剪,参数设置为“ILLUMINACLIP:2:30:10 SLIDINGWINDOW:5:20 LEADING:5 TRAILING:5 MINLEN:50”。清理后的读取数据使用Trinity(v2.8.5)(Grabherr et al. 2011)进行de novo组装,参数设置为“–jaccard_clip”。将每个样本的所有转录本使用GMAP(Wu和Watanabe 2005)与参考基因组‘Nijisseiki’对齐,进行参考转录本的过滤和去除。保留与参考基因组的相似度<85%且覆盖度<85%的序列,并定义为非参考转录本。这些非参考转录本与GenBank核苷酸数据库(2021年11月12日下载)及植物线粒体或叶绿体基因组使用blastn(Camacho et al. 2009)进行对齐,去除与绿色植物核基因组无关的序列,参数设置为“-evalue 1e–5 -best_hit_overhang 0.25 -max_target_seqs 10”。使用TransDecoder(v5.5.0)预测每个个体转录本的开放阅读框(ORF),然后使用CD-HIT(Li和Godzik 2006)去除冗余序列。
为获得每个个体的唯一非参考转录本,进行了两步重映射(附录B)。第一步,将非冗余的ORF与参考转录本合并作为索引。使用bowtie2(v2.4.4)(Langmead和Salzberg 2012)将对应的RNA-seq数据与索引对齐,使用bamdst(v1.0.9,https://github.com/shiquan/bamdst)计算覆盖度<100%的序列,并使用内部Python脚本(https://github.com/biolittleboy/Pan-transcriptome/blob/main/Coverage100_select.py)去除这些序列。第二步,将上一阶段剩余的新序列使用blastn与参考转录本对齐,参数为“-evalue 1e–5 -perc_identity 85 -qcov_hsp_perc 85”。将相似度≤85%、覆盖度≤85%且E值≤1e–5的序列定义为每个个体的非参考转录本。将每个样本中清理后的非参考转录本合并,使用CD-HIT去除冗余序列,参数设置为“-c 0.9”。最后,将最终的非冗余新转录本序列与梨的参考转录本合并,形成全转录组。
2.3. 基因注释和富集分析
基因功能注释通过使用DIAMOND(Buchfink et al. 2015)(E值≤1e–5)与Swiss-Prot(Jungo et al. 2012)和NR数据库(O’Leary et al. 2016)进行比对。转录因子(TFs)使用植物转录因子数据库(PlantTFDB)(Jin et al. 2017)进行注释。
基因本体(GO)注释使用InterProScan版本5.52-86.0(Jones et al. 2014)和InterPro数据库(Blum et al. 2021)。GO富集分析使用Fisher精确检验,通过topGO包实现(Alexa et al. 2006)。功能类别使用REVIGO(Supek et al. 2011),结果可视化使用CirGo(Kuznetsova et al. 2019)和ggplot2包(Ginestet 2011)。Kyoto基因与基因组百科全书(KEGG)通路和Pfam结构域注释使用EggNOG-mapper版本2.1.4(Cantalapiedra et al. 2021)。KEGG富集分析通过clusterProfiler包实现(Yu et al. 2012)。
2.4. ePAV识别与表征
在Trinity中使用align_and_estimate_abundance.pl脚本来计算与转录本对齐的序列片段数量。该脚本使用bowtie2将每个样本的读取与全转录本对齐,并使用samtools转换文件格式。批次效应通过使用sva包(Leek et al. 2012)进行去除。 FPKM值通过以下公式计算: FPKM = cDNA片段数 /(已对齐片段数(百万)× 转录本长度(kb))。 在超过95%的样本中表达(FPKM>1)的转录本定义为核心转录本,在1-95%样本中表达的转录本定义为外壳转录本,在<5%样本中表达的转录本定义为稀有转录本。对于给定的样本大小(范围为1-506),进行了10,000次随机抽样,并统计了抽样中表达的基因数。根据Tettelin等人(2008)的方法,取中位数来拟合幂律分布。
2.5. 群体结构
在IQ-TREE(Nguyen et al. 2015)中基于ePAV数据(以0/1表示)构建了最大似然(ML)系统发育树,并进行自助法检验(1,000次重复)。ML树在FigTree(v1.4.4)中进行可视化,并使用分支图形转换(FigTree)。主成分分析(PCA)使用来自scikit-learn库的“sklearn.decomposition.PCA”函数(About us — scikit-learn 1.7.0 documentation),基于相同的ePAV数据进行。
2.6. 土著品种与改良品种中的差异表达基因(DEGs)识别
我们计算了土著品种和改良品种中每个基因的表达比例(FPKM>1)。使用Fisher精确检验来确定土著品种与改良品种中每个基因的表达存在与缺失频率的显著性差异。然后,所有基因的原始P值经过假发现率(FDR)修正。使用DESeq2(Love et al. 2014)在P值<0.05和|log2(fold change)|≥2的标准下识别土著品种与改良品种之间的DEGs。基因表达的变异系数(CV)通过使用内部Python脚本(https://github.com/biolittleboy/Pan-transcriptome/blob/main/CV_cal.py)根据公式计算: CV = FPKM标准差 / 平均FPKM。
2.7. 共表达模块识别
使用WGCNA包(Langfelder和Horvath 2008)将表达的基因(平均FPKM>1)分配到具有相同表达模式的已识别模块。使用逐步网络构建和模块检测流程来识别梨的基因模块,参数参考了我们之前的研究(Wang et al. 2021)。共表达网络使用Cytoscape(v3.9.1)(Shannon et al. 2003)和Gephi(v0.9, Gephi - The Open Graph Viz Platform)进行可视化。在线共表达网络已在NDEx(Pillich et al. 2017)上共享,名称为“sugar_network”。
2.8. 表型相关性分析
从24个沙梨种质资源中收集了早期落叶病指数、果肉颜色和石细胞计数的表型数据,这些数据来自我们之前的研究(Zhang et al. 2021;Shan et al. 2023)。使用R进行Pearson相关性分析。
3. 结果
3.1. 沙梨全转录组
从包括果肉、果皮、叶子、花蕾和花粉在内的不同组织中生成并整理了506个沙梨样本的RNA-seq数据集,并采用去新组装和参考基因组对齐的策略构建了全转录组(附录A;图1-A)。在去除接头和低质量序列后,共获得了240亿个读取数据,每个样本的平均基因组大小为6,992,055,291个碱基对(图1-B;附录A)。每个样本的转录组进行了去新组装,总共获得了5700万个转录本,每个样本平均获得113,129个转录本(图1-C)。所有组装的转录本都与参考基因组‘Nijisseiki’(Shirasawa et al. 2021)进行了比对,以识别参考基因组中缺失的序列。每个样本约有90.67%的转录本可以与参考基因组对齐(图1-D)。这些对齐的转录本被排除在全转录组构建之外。经过去除污染物和叶绿体基因组序列的过滤后,最终获得了391万个原始新转录本,每个样本的平均原始新转录本数为7,725个。随后,使用TransDecoder预测了每个样本的最佳候选ORF。为了确认筛选出的最佳候选ORF序列的准确性,RNA-seq数据集重新映射到ORF和参考CDS的联合体上,使用bowtie2选择覆盖度为100%的ORF序列。接着,使用blastn进一步筛选和去除与参考转录组对齐的转录本。每个样本平均获得627个新转录本。然后将这些转录本合并去除冗余,最终得到111,868个非冗余新转录本。最后,将参考转录本与非冗余的新转录本合并,获得了沙梨的全转录组,共包含156,744个转录本。
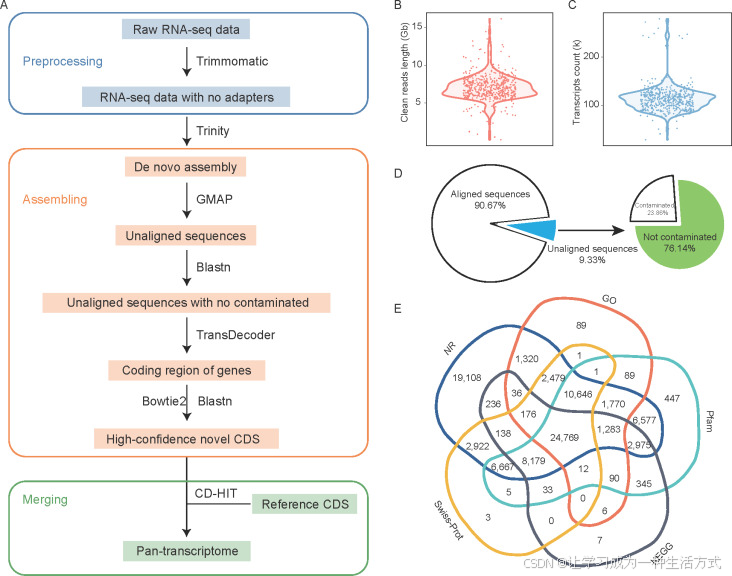
图1. 沙梨全转录组的构建 A, 构建流程,包括数据预处理、非参考转录本的组装和参考转录本与新转录本的合并。 B, 小提琴图显示每个样本的RNA-seq数据的清洁读取长度。 C, 小提琴图显示Trinity组装后的每个样本的转录本数量。 D, 去新组装转录本与参考基因组的比对比率统计,以及在非参考转录本中识别非污染序列。 E, Venn图展示了在NR、Swiss-Prot、KEGG、Pfam和GO数据库中注释的基因数量。
3.2. 新转录本的注释
新转录本的平均长度为547 bp,显著短于参考转录本的长度(1,248 bp)(图2-A)。分别使用GO、KEGG、NR、Swiss-Prot和Pfam数据库对42,767、38,285、89,281、56,031和63,888个新转录本进行了注释,其中24,769个转录本在这五个数据库中都有识别(图1-E;附录C)。在梨的全转录组中预测了转录因子,其中MYB、ERF、bHLH、NAC、C3H、WRKY、bZIP和C2H2家族占据了最高比例(图2-B)。新转录本中有1,401个转录因子,占所有转录因子的33%(附录D)。新转录本的平均变异系数(CV)为2.67,显著高于参考转录本的1.62(附录E)。这些结果表明,新转录本可能有助于沙梨的个体间差异。
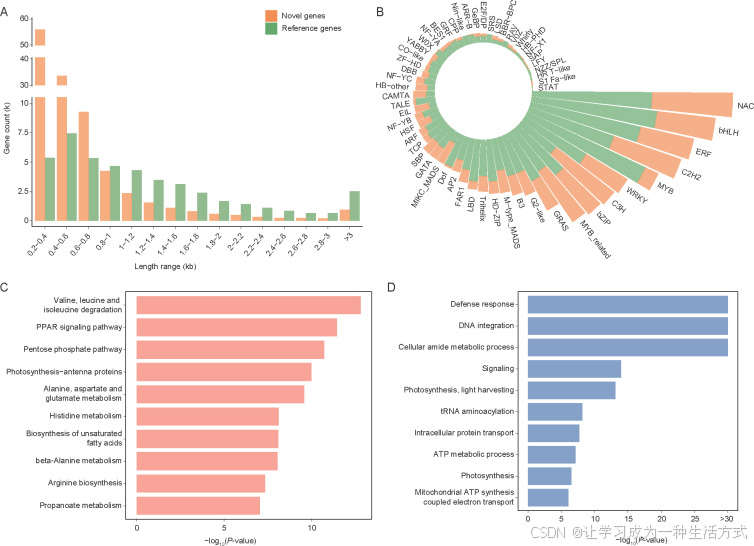
图2. 沙梨全转录组注释 A, 参考转录本与新转录本长度的比较。 B, 在全转录组中识别的转录因子及其参考转录本和新转录本的分布。 C, 新转录本的KEGG富集分析。 D, 新转录本的GO富集分析。
KEGG富集分析显示,新转录本在氨基酸代谢与降解、戊糖磷酸途径、不饱和脂肪酸的生物合成和光合作用等通路中显著富集(图2-C)。氨基酸、糖类和不饱和脂肪酸的代谢影响着香气的产生(Schwab et al. 2008),因此这些新转录本可能为果实品质性状的挖掘提供资源。GO富集分析显示,新转录本在多种生物过程中的富集显著,包括防御反应、DNA整合、细胞酰胺代谢过程、信号转导等(图2-D)。在新转录本中识别出了1,863个与防御反应相关的基因,占所有防御反应相关基因的74.6%,其中1,750个基因在Swiss-Prot数据库中有同源基因(附录C)。例如,Nonrefgene086797是AtMLP28的同源基因(附录F),AtMLP28是由昆虫害虫白蝶(Pieris rapae)卵特异诱导的免疫反应基因(Little et al. 2007)。此外,Nonrefgene006562与NtPR-4A(附录G)同源,NtPR-4A是与烟草花叶病毒(TMV)致病相关的基因,且该基因在感染TMV后表达显著增加(Friedrich et al. 1991)。这些结果表明,不同的沙梨个体可能已经在不同的环境条件下进化出了额外的防御反应基因。因此,全转录组的构建将通过应用这些抗性基因资源进一步促进梨的抗病虫育种进展。
3.3. 表达存在/缺失变异(ePAVs)发现与功能表征
ePAVs是群体转录组中的一种重要变异类型(Liu et al. 2020)。由于沙梨的高杂合度(0.89%)(Gao et al. 2021),选择了479个RNA-seq数据量较大的样本(>5 Gb)进行ePAVs的调用(附录A),在这些479个样本中有132,546个转录本被表示。全转录组转录本根据其出现频率进行了分类,得到了14.63%的核心转录本(≥95%)、42.76%的外壳转录本(1-95%)和42.61%的稀有转录本(≤1%)(图3-A和C)。稀有转录本占全转录组的近一半,表明沙梨群体中具有较高的基因表达多样性。通过对样本进行迭代随机抽样的方式,模型化全转录组转录本的数量,表明从479个样本构建的全转录组已接近饱和(图3-B)。该图显示,随着样本数量的增加,全转录组大小的增长减缓,表明大部分转录本ePAVs已经被捕捉。
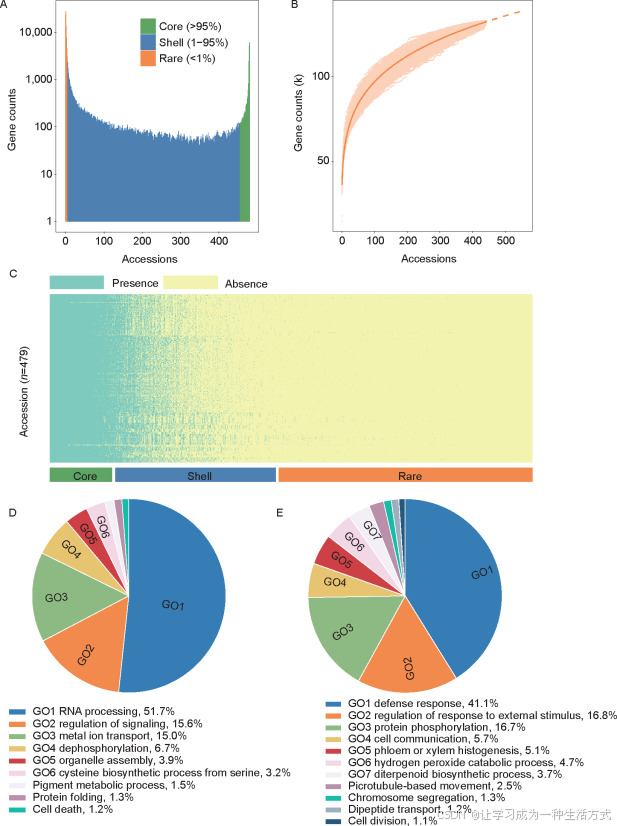
图3. 沙梨种质中的ePAV基因 A, P. pyrifolia全转录组基因的基因数量和出现频率。 B, 表示479个P. pyrifolia样本中全转录本数量增加的饱和曲线模型。 C, 热图显示479个P. pyrifolia样本中基因的表达存在与缺失情况。 D, CirGO可视化显示核心转录本中富集的GO术语(P<0.01)。 E, CirGO可视化显示外壳转录本中富集的GO术语(P<0.01)。
梨经历了两次全基因组重复(WGD)事件,并且在进化过程中发生了多拷贝基因丢失(Wu et al. 2013)。在对全转录组中的直系同源转录本进行聚类后,发现97%的核心转录本是重复的,而稀有转录本则具有最高比例的单拷贝基因(29%)(附录H)。这表明,大多数核心转录本在WGD之后保持了多拷贝状态,而稀有转录本在进化过程中更容易丧失多拷贝。核心转录本主要富集在一些关键功能中,如RNA处理、细胞表面受体信号通路、磷脂运输、去磷酸化、从丝氨酸合成半胱氨酸以及细胞器组装,因此它们在进化过程中可能对植物的生长和发育至关重要(图3-D)。外壳转录本则富集在防御反应、对外界刺激反应的调控、蛋白质磷酸化和细胞通讯等生物过程中(图3-E)。这突出了与抗性和环境适应相关的基因并不需要在每个品种中都有,但它们分布在不同的样本中。
3.4. 基于ePAV的沙梨亚群系统发育关系
沙梨在Pyrus属中具有最高的核苷酸多样性(Wu et al. 2018),并且拥有大量的地方品种和改良品种。在479个样本中,共表达了126,413个转录本,其中74.8%来自新转录本(表达率<100%),这些转录本用于构建系统发育树、主成分分析(PCA)和后续分析。这些样本代表了174个地方品种和215个改良品种(附录A)。
通过ePAV数据可以区分地方品种和改良品种(图4-A)。梨经历了较弱的驯化,一些地方品种和改良品种未表现出明显的遗传差异,这导致一些种质在系统发育树中聚集在一起(图4-A)。根据PCA结果,改良品种被分为改良I组和改良II组(图4-B)。改良I组主要包括来自日本和韩国的种质(第1组)及其杂交后代作为亲本;改良II组对应来自中国的改良品种(第2组)。地方品种被分为地方品种I组和地方品种II组(图4-B)。梨的传播途径有两个,分别是从中国西南部经过长江流域向东传播,以及通过珠江流域向南传播(Chen et al. 2022)。来自梨原产地附近的四川省和浙江、广东、广西等省份的样本被归为第5组。
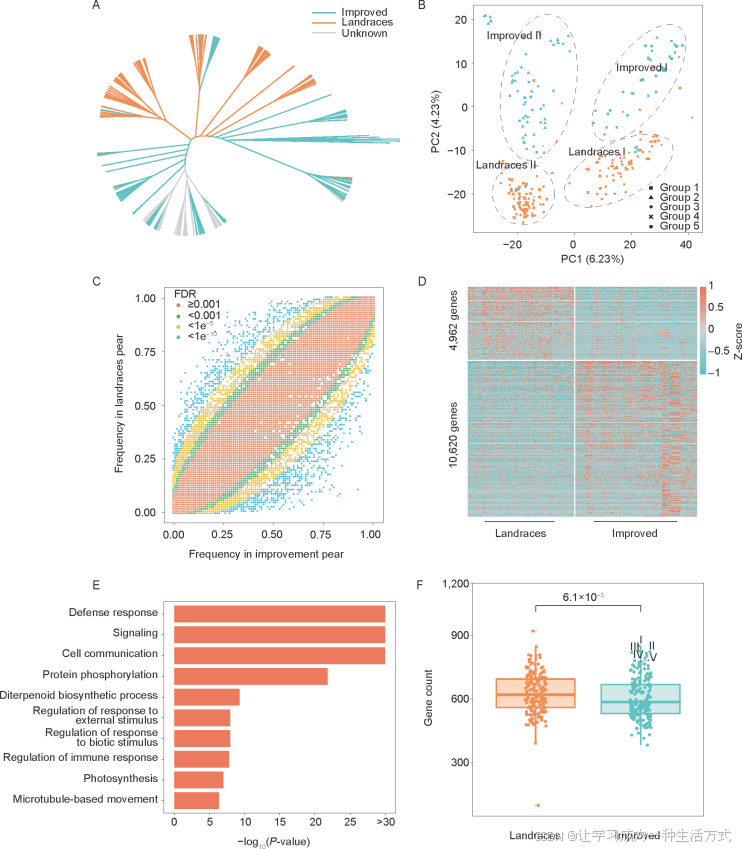
图 4. 梨(*Pyrus pyrifolia*)的群体结构和通过ePVAs计算的改良过程中的转录组选择
A:基于ePAV数据的系统发育树。橙色表示地方品种组;天蓝色表示改良品种组;浅灰色表示分类不清晰的样本。 B:基于ePAV数据的479个样本的主成分分析。方块(组1)表示来自日本和韩国的改良样本;三角形(组2)表示在中国培育的国内外衍生品种;菱形(组3)表示来自中国湖北、贵州和云南的地方品种样本;十字(组4)表示来自中国湖南、江西和福建的地方品种样本;圆圈(组5)表示来自中国四川、广东、广西和浙江的地方品种样本。 C:显示地方品种和改良品种中每个基因的表达频率的散点图。 D:地方品种和改良品种中的差异表达基因(DEGs)热图。高表达水平用红色表示,低表达水平用蓝色表示。 E:新转录本的GO富集分析。 F:地方品种和改良品种中防御反应基因数量的箱线图。显著性通过Wilcoxon检验计算。I,荣山梨;II,温环梨;III,早熟梨;IV,脆杏梨;V,脆冠梨。
在流域的尽头,所有样本聚集在地方品种I和II中(图 4-B;附录 I)。来自中国云南、贵州和湖北省的样本(组3)聚集在地方品种I中。来自中国湖南、江西和福建省的样本(组4)沿长江流域聚集在地方品种II中。通过479个沙梨样本的ePAVs,出现了两类沙梨地方品种。这两组沙梨种质经历了不同的传播过程,最终在长江和珠江流域的尽头汇聚。
3.5. 改良后抗性基因表达较低
大多数为了提高产量而选择的现代作物品种对病原体的抗性较差(Deng等,2020)。梨的改良主要发生在过去十年,而且在改良过程中对抗性应激的关注较少(Li等,2019)。地方品种和改良梨品种之间的群体结构差异明显(图 4-A和B),并且地方品种与改良品种之间的RNA-seq比对率存在差异(附录J和K)。
通过比较地方品种和改良品种是否表达基因,确定了改良过程中选择的基因。对于每个比较,表达频率在地方品种和改良品种之间存在显著差异(P<0.001)的转录本被确定为选择的转录本,并且这些转录本在防御反应的生物过程注释中显著富集(图 4-C;附录L)。在改良品种中频率较高的转录本被认为处于正选择状态,而频率较低的则被认为处于负选择状态。总的来说,从改良过程中发现了4,139个和4,430个正负选择转录本(附录M)。在选择的转录本中,共鉴定出396个防御反应基因,其中82.3%来自新转录本。正选择和负选择下分别有151(3.65%)和245(5.53%)个防御反应转录本。根据Fisher确切检验,正负选择下防御反应基因的数量存在显著差异(P=3.6e–05)。这表明在改良过程中,许多防御反应基因被负向选择。
为了评估表达水平的差异,共鉴定了15,582个差异表达基因(DEGs),其中8,892个来自新转录本(附录N)。地方品种中有4,962个高表达基因,而改良品种中有10,620个高表达基因(图 4-D)。这些DEGs在防御反应的生物过程也表现出富集(图 4-E)。地方品种中的防御反应基因高表达的数量显著高于改良品种(P<2.2e–16)。这与地方品种比改良品种具有更强抗病性这一认识一致。
通过上述两种方法发现了18,095个基因(附录O)。基于构建的泛转录组,更多的基因在地方品种和改良品种之间表现出差异表达。与改良过程相关的防御反应基因在表达数量和水平上均有所下降,这些选择的防御反应相关基因主要来自新转录本。在每个样本中计算防御反应基因的数量,地方品种中显著更多的防御反应基因相比于改良品种(图 4-F)。地方品种II中的抗病基因资源比地方品种I更多(附录P)。中国培育的改良品种相比于韩国和日本培育的品种有更多的防御反应基因,这可能归因于丰富的地方品种种质资源。尽管改良品种的抗病基因总体上少于地方品种,但仍有一些优良品种具备较强的抗病性。在改良品种中,防御反应基因最多的品种为荣山梨、温环梨、早熟梨、脆杏梨和脆冠梨(图 4-F;附录Q)。将这些品种作为杂交亲本进行品种改良,可能有助于提高后代的抗性和环境适应性。
3.6. 不同梨性状之间的相关性
基因共表达网络可用于识别功能基因模块(Ruan等,2010),并通过特征基因与性状进行关联(Langfelder和Horvath,2008)。候选共表达模块是基于44,139个基因(平均FPKM>1)通过WGCNA识别的,其中42.1%来自新转录本。通过样本聚类分析去除了两个异常样本(A167_1和SRR12614248),剩余的477个样本用于后续分析(附录R)。通过大约无标度拓扑法计算得到软阈值β为4(附录S)。总共有41,204个基因被分为25个模块(M1–M25)(图 5-A),其余的2,935个基因被视为异常值。五个模块(M2–M3和M20–M22)与基本功能(如mRNA处理)相关,这些模块中的参考转录本数量大于新转录本数量(附录T)。模块M17和M23分别富集于“花发育的负调控”和“长日照期调节、开花的正调控”,因此这两个模块可能与花发育相关(附录T)。
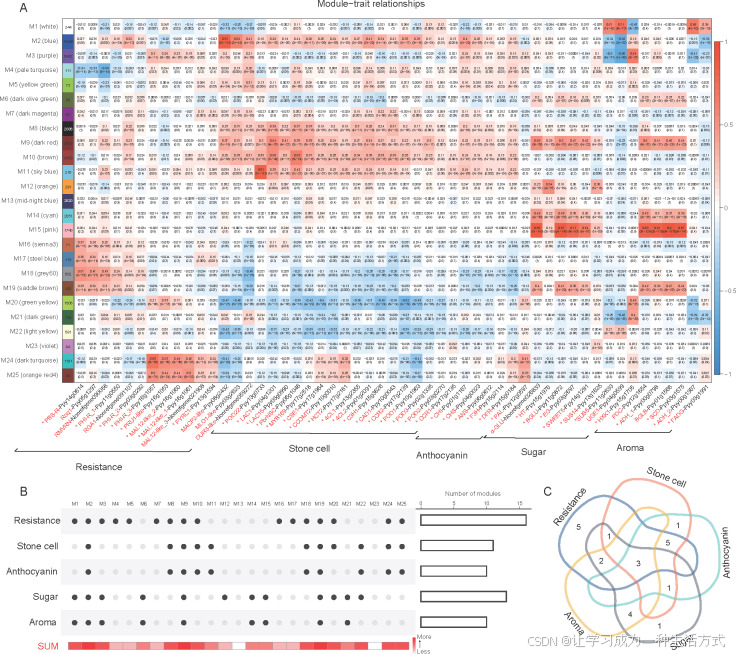
图 5. 抗性和果实质量性状相关基因的加权基因共表达网络分析(WGCNA)
A:模块与性状的关系。行代表模块M1–M25,左侧的数字表示每个模块中包含的基因数量。每列对应影响梨的抗性和果实质量的基因。星号(*)表示在梨中已确认功能的基因。每个行列交点处包含模块与特征基因之间的相关系数和P值。红色表示正相关,蓝色表示负相关。 B:与抗性和果实质量性状相关模块的统计。黑色圆圈表示相关,灰色圆圈表示不相关。右侧的柱状图表示相关模块的数量。在下方的热图中,较深的红色表示模块与更多性状相关。 C:与抗性和果实质量性状相关模块的韦恩图。
当梨的防御机制被触发时,病理相关基因和防御相关基因如过氧化物酶(POD)的表达水平显著增加(Sun等,2018;Yan等,2018)。作为木质素合成的关键酶,POD对梨中石细胞的形成至关重要(Xue等,2019a)。然而,石细胞的形成是否与梨的抗病性相关仍然未知。因此,我们选择了影响石细胞形成的基因,包括POD和漆酶(LAC)、转录因子MYB和NSC,以及影响梨抗性的基因,如PR基因和防御反应相关基因,来与共表达模块关联(图5-A)。与抗性相关的16个模块(P≤0.01)在防御反应(M4, M7, M16, M18–M19, M24)、系统获得性抗性(M9-M10)、先天免疫反应(M25)、过氧化物酶体组织(M5)和响应高光强(M8)等生物过程中富集(图5-A和B;附录T)。与石细胞形成相关的11个模块(P≤0.01)在木质素分解过程(M10–M11)、半纤维素代谢过程(M9)、细胞壁修饰(M25)、微管基础的运动(M2, M9)、植物激素响应(M8, M10, M24)、光合作用(M8)和木聚糖生物合成过程(M10)等生物过程中富集(图5-A和B;附录T)。九个模块与石细胞形成和抗性共同相关(图5-B),其中五个模块(M8–M10, M24–M25)共同富集了石细胞形成和抗性相关的功能(附录T)。相关性分析显示,早期落叶病指数与石细胞数量之间存在正相关(附录U和V),表明随着病情的进展,石细胞数量显著增加(P<0.05)。这表明石细胞的形成可能与梨的抗病性相关。
酚丙烷生物合成途径控制木质素和花青素的合成,且它影响梨果实石细胞的形成和色素的沉积(Xue等,2019b;Liu等,2021)。共发现10个与花青素合成相关的模块(P≤0.01)(图5-A)。有趣的是,这10个模块与石细胞形成相关的模块完全重合,且其中8个模块也与梨的抗病性相关(图5-B)。相关性分析显示,早期落叶病指数与果肉颜色之间存在正相关(附录U和V),表明随着病情的进展,花青素含量显著增加(P<0.05)。这表明花青素的合成也可能与梨的抗病性相关。
梨的一些香气成分来源于由糖类底物合成的二次代谢物(Bood和Zabetakis,2002)。通过使用香气和糖合成基因的表达水平进行表型分析,共发现9个模块与香气和糖合成共同相关(图5-B)。这些模块的基因富集在葡萄糖代谢过程(M1)、D-氨基酸分解过程(M1)、1,3-β-D-葡聚糖合成过程(M21)、脂肪酸合成过程(M14)、支链氨基酸代谢过程(M19)和二萜类生物合成过程(M9)等注释中。模块M2、M9和M19与抗性、石细胞形成、花青素、糖和香气合成相关(图5-C),这表明这三个模块中的基因可能对育种具有重要意义。
3.7. 糖合成通路中结构基因和转录因子的共表达网络
糖的组成和数量是影响梨果实质量的重要因素。糖合成涉及淀粉和蔗糖代谢通路(map00500)、半乳糖代谢(map00052)、果糖和甘露糖代谢(map00051)、戊糖和葡萄糖醛酸相互转化(map00040)以及氨基糖和核糖糖代谢(map00520)等。我们的分析发现糖合成网络中有56种酶(Wu等,2018;Li等,2022),共涉及1,090个与糖合成相关的基因,其中717个来自新转录本(图6-A)。新转录本中包含糖相关基因,为未来梨果实质量研究提供了额外的数据资源。
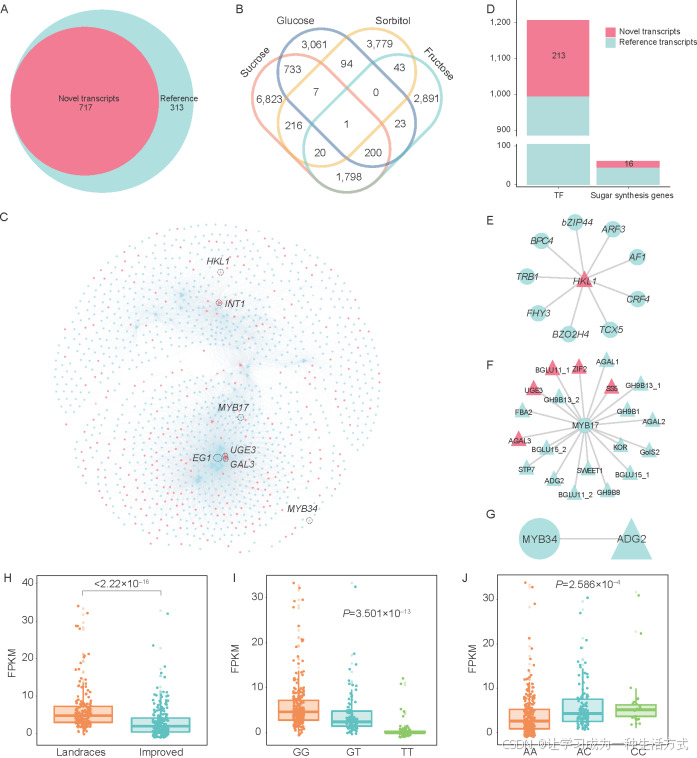
图 6. 糖合成相关基因和转录因子的共表达分析
A:参考转录本(蓝色)和新转录本(粉红色)中的糖合成相关基因。 B:与蔗糖、葡萄糖、山梨醇和果糖含量相关的基因的韦恩图。 C:参与糖合成和转录因子的基因网络。参考转录本中的基因以蓝色显示,新转录本中的基因以粉红色显示。糖合成通路中的基因用三角形表示,转录因子(TFs)用圆圈表示。 D:共表达网络中糖合成相关基因和转录因子的数量柱状图。 E:HKL1基因和转录因子的共表达关系。 F:MYB17与糖合成相关基因的共表达关系。 G:MYB34与ADG2(葡萄糖-1-磷酸腺苷转移酶)的共表达。 H:地方品种和改良品种中HKL1基因表达水平的箱线图。 I和J:HKL1中的两个SNP,Nonrefgene027455_273(I)和Nonrefgene027455_171(J),显示出在具有三种不同基因型的种质间HKL1表达水平的差异(t检验)。
通过基因表达与20个样本的糖含量之间的相关性分析,共发现了泛转录组中的19,689个基因与果糖(4,976个)、山梨醇(4,160个)、葡萄糖(4,119个)和蔗糖(9,798个)含量显著相关(P<0.05)(图6-B;附录W)。其中有114个基因被注释为糖合成相关基因,61个基因位于479个样本的共表达网络中(附录W和X)。糖合成相关基因与转录因子的共表达关系被提取自共表达网络(图6-C),1,268个糖相关基因和转录因子有共表达关系,其中229个来自新转录本(图6-D;附录X)。
Ppy11g2632.1(EG1,内切葡聚糖酶)是网络中连接最多的结构基因,在网络中有489个节点。GAL(α-半乳糖苷酶)、INT(肌醇转运蛋白)和UGE(UDP-葡萄糖4-表异构酶)在糖的合成和运输中发挥作用(Nicolai等,2006;Li等,2015;Arunraj等,2020)。来自新转录本的Nonrefgene011861.1(GAL3)、Nonrefgene007701.1(INT1)和Nonrefgene055723.1(UGE3)分别与238、229和205个转录因子共表达(图6-C)。HXK1可以调节果实中的糖含量,并影响梨的生长发育(Zhao等,2019)。作为己糖激酶基因注释的Nonrefgene027455.1(HKL1)出现在网络中,并与九个转录因子共表达(图6-E)。在网络中,共识别了162个MYB转录因子,其中MYB17与20个糖合成相关基因有共表达关系,其中5个基因来自非参考转录组(图6-F)。值得注意的是,MYB34与Ppy16g1265.1(ADG2,葡萄糖-1-磷酸腺苷转移酶)共表达(图6-G)。
在共表达网络中,有16个(26.23%)糖相关基因的表达水平在梨的改良过程中发生了变化。其中,Nonrefgene027455.1(HKL1,己糖激酶)在地方品种中的表达水平显著高于改良品种(图6-H;附录N)。我们在HKL1基因中发现了两个SNP,包括与Nonrefgene027455_273对应的GG等位基因,主要存在于地方品种中,而GT/TT等位基因主要存在于改良品种中(附录Y)。与Nonrefgene027455_171对应的AA等位基因主要存在于改良品种中,而AC/CC等位基因主要存在于地方品种中(附录Z)。这两个SNP显著影响了HKL1基因的表达(图6-I和J),这种联系可能是地方品种和改良品种之间HKL1基因表达差异的原因。Nonrefgene079849.1(SS5,淀粉合成酶)和Nonrefgene009327.1(BGLU11_1,β-葡萄糖苷酶)在改良品种中的表达水平显著高于地方品种(附录N),并且它们与MYB17共表达(图6-F)。这些新转录本可能是沙梨改良过程中甜度变化的原因之一。
4. 讨论
本研究组装了梨的第一个泛转录组,包括了在参考基因组“ Nijisseiki”中未发现的111,868个新转录本。沙梨的泛转录组中新转录本的数量超过了玉米(8,681个)(Hirsch等,2014)、番茄(7,181个)(Liu等,2020)和茶树(4,940个)(Kong等,2022),但少于大麦(289,697个)(Ma等,2019)。沙梨中新转录本的平均长度(547 bp)短于参考转录本(1,248 bp),这与玉米中的发现一致(Hirsch等,2014)。此外,我们还观察到沙梨中新转录本的表达水平较低(附录AA)。低表达水平的基因通常具有较短的转录本长度,这可能会引入对这些转录本的检测偏差(Oshlack和Wakefield,2009)。Pyrus中的新转录本的长度和表达水平表现出类似的趋势。这些新转录本在响应生物和非生物胁迫的基因中富集,并且作为非必需基因(<95%)在沙梨种群中分布,可能用于适应不同的环境条件。
由于基因表达的时空特异性,通过RNA-seq数据构建的泛转录组不能像基因组重测序数据构建的泛基因组那样全面地包含物种多样性信息。然而,泛基因组中的许多基因是未转录的(Zhao等,2018;Gao等,2019)。泛转录组可以提供时空基因表达的证据,这对于评估潜在的基因功能非常有价值。本研究并未观察到来自相同组织的沙梨样本的聚类(附录AB)。来自果肉组织以外的组织的RNA-seq数据有限,阻碍了不同组织之间差异的比较。在沙梨的更多组织类型和更多生长阶段进行转录组测序将增强泛转录组的完整性,并推进沙梨种群比较基因组学研究。沙梨的高遗传多样性对构建具有代表性的泛转录组构成了挑战。然而,通过识别ePAVs和预测建模,我们发现我们的泛转录组捕获了大多数基因表达的多样性。
沙梨的多样性和分布可能使不同个体中出现更多的ePAVs。在番茄的泛基因组中,核心基因的比例高达74.2%(Gao等,2019),而在番茄的泛转录组中,核心转录本的比例较低,仅有42.0%的基因在所有样本中表达(Liu等,2020)。在与梨密切相关的另一个蔷薇科物种——苹果(Malus domestica cv. Gala)中,核心基因仅占22.21%,而外壳基因占65%,稀有基因占12.75%(Sun等,2020)。相比之下,梨的泛转录组中稀有转录本的比例显著更高,这表明基因表达的时空特异性导致了核心基因和外壳基因的表达差异,因此,不同梨品种之间外壳基因和稀有转录本的表达差异可能与相应的表型变化和环境适应性相关。沙梨泛转录组中的稀有转录本包括52,361个(92.71%)新转录本。约一半的新转录本仅在少数样本中表达,表明新转录本可以为研究梨的个体间差异提供数据支持。
在长期的作物育种过程中,研究人员发现不同性状之间存在统计相关性(Cramer和Wehner,2000;do Rego等,2011)。例如,梨果实中糖类和香气化合物的合成存在相关性(Li等,2018)。梨石细胞形成相关基因,以过氧化物酶(POD)为代表,也参与梨的抗病性。POD基因对植物抵抗病原微生物非常重要(Sun等,2018;Yan等,2018),它们也参与梨的木质素合成和石细胞聚集(Cao等,2016)。然而,石细胞的形成是否与梨的抗病性相关尚不明确。在梨的育种过程中,抗性和果实质量无法有效平衡。我们发现,在进行香气和糖含量的选择育种时,抗性被排除在外;而在选择石细胞性状时,病害抗性和果色也被同时选择,但未确定为正向选择或负向选择。本研究中果实质量性状与抗病性之间的相关性,可以为抗病育种过程中亲本选择提供参考。
现代梨育种的目标是实现优良的果实质量、高水平的抗病性、自交兼容性以及降低收获期间的劳动成本(Saito,2016)。在驯化和改良过程中,育种者倾向于更多关注果实质量性状,如果实大小、甜度、石细胞含量、香气和颜色。在番茄中过表达AtMYB12显著降低了主要糖类的含量(Zhang等,2015)。在梨的泛转录组中发现了162个MYB转录因子,其中MYB17与AtMYB12同源,但尚未有关于MYB17调节梨糖合成的报道。CiMYB17是与梨中的MYB34同源的基因,参与菊苣中的果聚糖合成和降解(Wei等,2017)。在我们之前的研究中,我们观察到MYB34的表达水平和果糖含量在梨果实发育过程中呈持续上升趋势(Li等,2016;Zhao等,2019)。我们构建的结构基因与糖合成相关转录因子的共表达网络,结合参考转录本和新转录本,能为挖掘品质性状基因提供参考。
驯化和改良过程还导致了遗传多样性的减少,尤其是在抗病性方面。火疫病、梨斑病、黑斑病和梨蚜是梨栽培过程中遇到的主要病害(Li等,2022)。我们对不同样本的ePAVs分析表明,梨园香、梨园黄和梨园城托在沙梨地方品种中含有更多的防御反应基因。在改良品种中,脆冠梨由于其广泛的适应性和优良的果实质量,包括薄皮、大果、脆爽、早熟以及甜美清新的口感,成为中国流行的梨品种(Wang等,2015)。我们发现脆冠梨表达的防御反应基因数量低于荣山梨、温环梨、早熟梨和脆杏梨。使用上述样本可以进一步提高梨的抗病性和环境适应性,在未来的梨育种中,通过挖掘泛转录组中的新转录本,可以为果实质量育种和抗性育种提供新资源。
5. 结论
总之,我们构建了第一个果树作物的泛转录组,并建立了梨抗病性和果实质量育种的数据库资源。我们采用创新的转录组学方法揭示了梨个体间复杂的表达变异,从而为了解多年生果树作物中的遗传多样性提供了宝贵的见解。