基于 GWAS 的群体遗传分析将 bZIP29 确定为玉米中的异种基因
https://www.sciencedirect.com/science/article/pii/S2590346225000513?via%3Dihub
摘要
了解异质基因在杂种优势形成中的作用对于推进杂交育种至关重要。我们分析了玉米杂交种群体的株高(PH)、穗高(EH)和转录组数据。全基因组关联研究(GWAS)表明,数量性状位点(QTL)的显性效应在杂交性状和中亲优势中起着重要作用。通过整合GWAS、表达GWAS(eGWAS)和模块eGWAS分析,我们优先确定了六个候选异质基因,这些基因位于六个QTL之下,其中包括一个跨越bZIP29基因的QTL。在杂交种群体中,bZIP29表现出对杂交性状和中亲优势的加性表达和显性效应,其有利等位基因与PH和EH呈正相关。在转基因与野生型系杂交的杂交种中,bZIP29表现出显性或超显性模式,这取决于其表达水平。通过tsCUT&Tag测定法发现,bZIP29蛋白直接与由其相关表达QTL(eQTL)调控的基因以及由其相关模块eQTL(meQTL)调控的表达模块中的六个基因结合。涉及bZIP29的调控网络在杂交亚群体中比在亲本群体中更为广泛。本研究为支撑杂交玉米强健生长的关键异质基因和网络提供了见解。
“异质基因”(heterotic genes)是指在杂交后代中表现出显著优势的基因。这些基因通常与杂种优势(heterosis)相关,即杂交后代在生长、发育或产量等方面表现出优于其亲本的特性。杂种优势是杂交育种中一个重要的现象,广泛应用于农业和园艺领域,以提高作物的产量和抗逆性。
引言
通过两个纯合系杂交产生的杂交种,通常会表现出比其亲本系更优越的农艺性状。提升杂交种的性能是提高作物产量的关键策略(Labroo 等,2021)。未来的育种工作需要全面理解多样化作物性状的分子调控机制,尤其是与农艺性状相关联的数量性状位点(QTL)和基因(Wallace 等,2018)。大多数旨在阐明基因功能的研究都以自交系为遗传背景,但这些研究对于理解杂交群体中的表型变异的参考价值有限,因为决定性状表现的QTL和遗传效应在自交系和杂交系之间往往存在差异(Guo 等,2014;Ma 等,2022)。因此,研究杂交性能的分子基础和调控机制对于优化杂种优势的利用至关重要。
传统的基于连锁的双亲群体研究(例如永生化F₂群体)已在多种作物物种中鉴定出与杂交性能相关联的数量性状位点(QTL),包括水稻(Hua 等,2002;Zhou 等,2012)、玉米(Tang 等,2010;Guo 等,2014)和棉花(Liu 等,2011)。值得注意的是,当分析不同的性状或群体时,鉴定出的与杂交性能和中亲优势(MPH)相关的QTL存在差异(Fievet 等,2018;Xiao 等,2021)。双亲群体固有地提供较低的定位分辨率,这使得鉴定支撑杂交性能的候选基因变得复杂(Liu 等,2020)。作为一种替代方法,通过杂交多种自交系产生的杂交群体已被用于剖析杂交性能(Seymour 等,2016)。由于这些群体具有较高的遗传多样性和定位分辨率,它们显著推进了我们对杂交性能遗传调控的理解(Huang 等,2015;Seymour 等,2016;Xiao 等,2021)。这些分析扩展了关于杂种优势的理论(Jiang 等,2017;Bonnafous 等,2018;Xie 等,2022),并揭示了杂交性能和MPH具有复杂的遗传结构,由许多基因及其相互作用塑造(Jiang 等,2017)。尽管广泛的遗传分析可以鉴定出与杂种优势或杂交性能相关的QTL,但这些研究对于揭示异质QTL的调控模式仅提供了有限的见解。
系统遗传学方法是研究动物和植物中支撑性状的基因、通路和网络的强大工具(Civelek 和 Lusis,2014)。在这些方法中,表达QTL(eQTL)定位被广泛用于建立基因组变异与基因表达之间的全基因组调控关系(Fu 等,2013;Li 等,2013b)。影响多个基因表达的基因组位点通常被定义为顺式作用热点,它们可能在调控目标性状的调控网络中发挥核心作用(Li 等,2020;Tan 等,2022)。然而,鉴定顺式作用关联可能具有挑战性,因为它们通常对来自生物学和技术变异以及组织和发育阶段差异的混杂因素敏感(Kolberg 等,2020)。转录组数据可以通过降维分析被组织成数十个模块(Rotival 等,2011;Cote 等,2022)。最近的研究表明,模块eQTL(meQTL)定位——将共表达模块的特征值视为复合性状——可以增加检测顺式调控热点的统计功效(Rotival 等,2011;Kolberg 等,2020;Sun 等,2023),并优先鉴定与性状表现相关的候选基因(Momozawa 等,2018;Tang 等,2021)。植物中的系统遗传学研究主要集中在建立自交群体中基因型、基因表达和表型之间的关系(Fu 等,2013;Tang 等,2021),对杂交群体的使用则较为有限。
关于杂种优势的形成机制,提出了包括显性假说和超显性假说在内的多种假设(Hochholdinger 和 Baldauf,2018)。根据显性假说,有利等位基因和不利等位基因在杂交系中对植物生长表现出显性和隐性效应(Jones,1917)。East(1936)提出,异质等位基因之间的互作会导致超显性效应。然而,将等位基因表达与异质效应联系起来的证据仍然有限。Springer 和 Stupar(2007)提出,在自交系中,基因表达水平可能被固定在最优范围之上或之下,而在杂交系中,中亲表达水平可能将基因表达水平调整到最优范围内。因此,杂交种可能由于中亲表达水平而表现出显性或超显性优势。尽管如此,支持这一模型的实验性证据仍然有限。
为了鉴定与杂交性能和杂种优势相关的异质基因和调控网络,我们基于北卡罗来纳II(NCII)设计构建了一个玉米杂交群体。由于株高(PH)和穗高(EH)在玉米中表现出强烈的杂种优势和高遗传力(Hu 等,2017;Li 等,2021b),这些性状非常适合用于研究杂交性能和杂种优势。此外,该群体的RNA表达谱为研究PH和EH的调控机制提供了宝贵的见解。将RNA表达数据与遗传定位相结合,有助于揭示杂种优势的分子调控机制。具体而言,本研究包括以下目标:(1)开展全基因组关联研究(GWAS),揭示PH和EH杂交性能的遗传基础;(2)通过表达GWAS(eGWAS)和模块eGWAS(meGWAS)分析,鉴定QTL背后的候选基因及其调控网络;(3)揭示异质基因bZIP29的表达如何影响杂交系中的杂种优势。鉴定出的异质基因及其相关网络有助于我们理解杂种优势的机制。
结果
杂交群体中株高(PH)和穗高(EH)的数量性状位点(QTL)鉴定
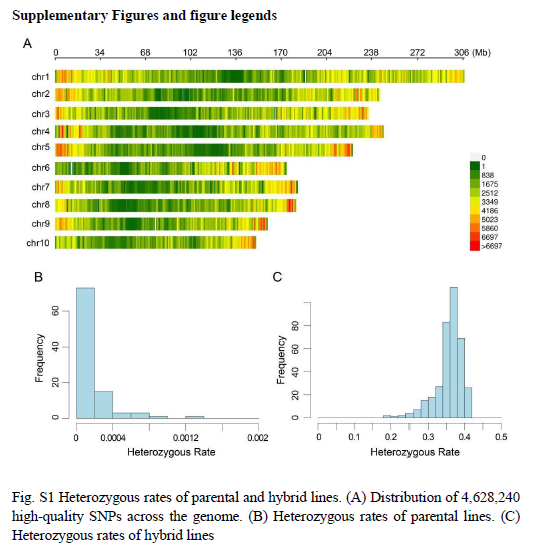
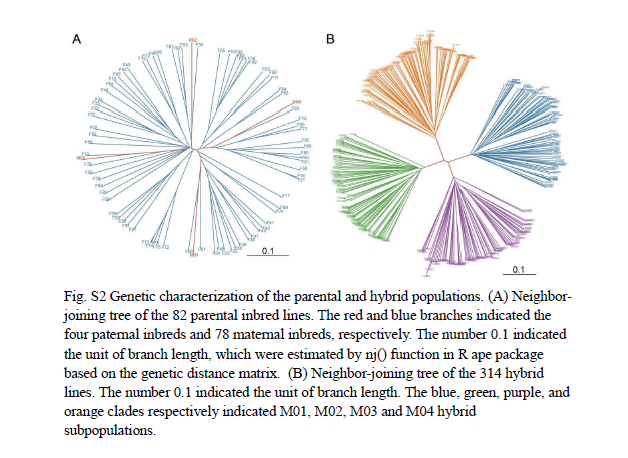
我们通过将78个母本系与4个父本系杂交,构建了一个北卡罗来纳II(NCII)设计的杂交群体,共产生314个杂交系(包括通过亲本系杂交获得的两个杂交系)。来自82个亲本系的全基因组重测序数据使我们能够鉴定出4,628,240个高质量的单核苷酸多态性(SNPs)(补充图1A)。亲本系的杂合率低于0.002(补充图1B),而杂交系的杂合率介于0.19到0.42之间(补充图1C)。系统发育分析显示亲本系的分布相对平衡,没有高度分化的类群,并且将四个雄性亲本分别置于不同的分支中(补充图2A)。杂交系被分为四个不同的类群,每个类群对应于四个父本自交系中的一个(补充图2B)。
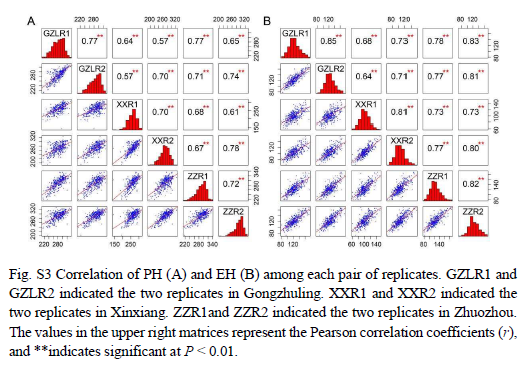
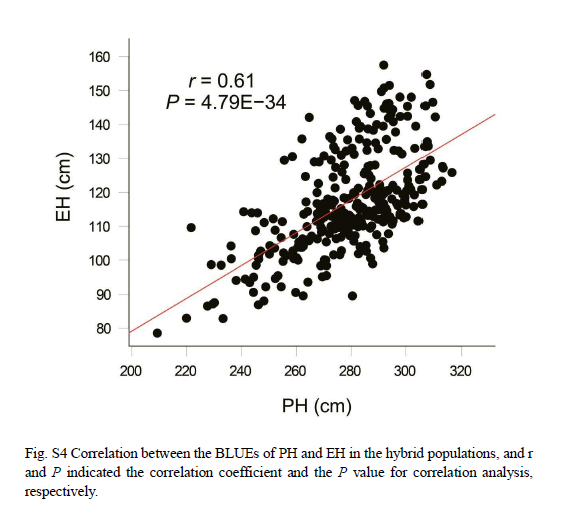
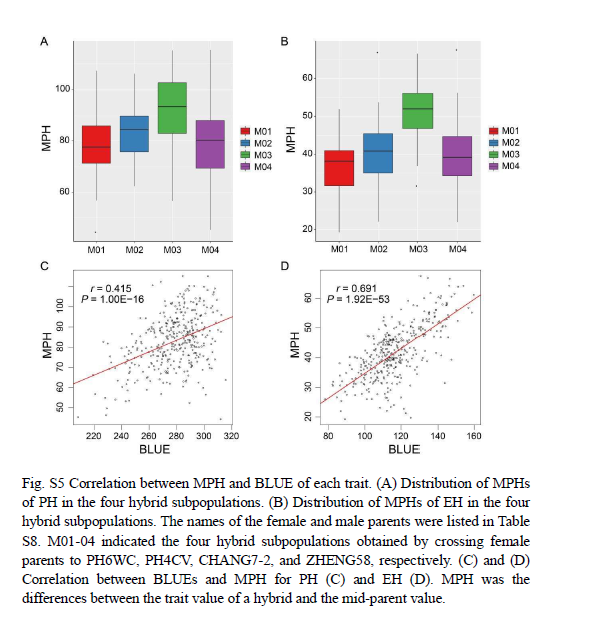
表型分析显示,不同环境内和环境间的重复之间存在强相关性(补充图3A和3B),株高(PH)和穗高(EH)之间也存在强相关性(补充图4),这证实了表型数据的可靠性。杂交系的表型与中亲表型的比较表明,所有杂交系在这两个性状上都表现出强烈的杂种优势(图1A),其中株高的中亲优势(MPH)值范围为45厘米到116厘米(补充图5A),穗高的MPH值范围为19厘米到68厘米(补充图5B)。株高(PH)和穗高(EH)的广义遗传力(H²)为0.96,而MPH的广义遗传力为0.70(株高)和0.77(穗高)(图1B),这表明杂交系在不同环境中表现出稳定的性能。
我们专注于杂交性能的遗传剖析,而不是中亲优势(MPH),因为杂交性能的广义遗传力(H²)高于其衍生的MPH(Li等,2021b)(图1B),并且这两个性状及其衍生的MPH值之间存在强相关性(补充图5C和5D)。我们使用了一个包含每个SNP的加性效应和显性效应的混合线性模型,共涉及2,324,652个SNP,用于全基因组关联研究(GWAS),并将其称为加性-显性(AD)模型(见方法)。AD模型能够有效减少由于群体结构和隐秘亲缘关系导致的潜在Ⅰ型错误(补充图6A-6D)。我们鉴定出11个加性QTL和41个显性QTL与株高(PH)相关,以及21个加性QTL和6个显性QTL与穗高(EH)相关(补充图7A-7D和补充表1)。对PH和EH的QTL物理位置进行比较发现,1个加性QTL和3个显性QTL与这两个性状均相关(图1C和补充表1)。显性QTL解释的表型变异(PVE)显著高于加性QTL对这两个性状的解释程度(图1D)。随后,我们在QTL区域内筛选了已注释的基因或已报道与植物生长或发育相关的同源基因,这些基因被认为是候选基因。在与PH相关的QTL中,有6个加性QTL和15个显性QTL,以及在与EH相关的QTL中,有8个加性QTL和4个显性QTL包含这些与植物生长或发育相关的候选基因(补充图7A-7D和补充表1)。
“有利”评分(代表有利QTL基因型的累积效应)与这两个性状的杂交性能显著相关(图1E和1F),这支持了这些QTL的可靠性。大多数显性QTL的显性/加性(d/a)比值(显性程度)超过1.0,而所有加性QTL的d/a值范围在-1.0到1.0之间(补充图8A和8B),这与这些QTL的遗传效应一致(补充表1)。这些结果表明,加性-显性(AD)模型能够可靠地区分加性QTL和显性QTL。此外,具有较高d/a值的QTL倾向于对表型变异的贡献更为显著(补充图8A和8B)。这些QTL的遗传效应进一步得到了不同基因型组之间表型比较的支持。具体而言,对于加性QTL,所有三个基因型组之间的杂交性能均存在显著差异(补充图9A和9B)。相比之下,对于显性QTL,具有杂合基因型的杂交种通常表现出比具有纯合不利等位基因的杂交种更高的性状值,但与具有纯合有利等位基因的杂交种之间没有显著差异(补充图9C和9D)。在显性QTL上具有杂合基因型的杂交种的中亲优势(MPH)值通常高于具有纯合等位基因的杂交种,而这种模式在加性QTL中并未观察到(补充图10)。
全基因组鉴定eQTL和eQTL热点
为了探索杂交性能的调控机制,我们对28,706个表达基因进行了表达全基因组关联研究(eGWAS),以全基因组扫描eQTL。我们使用了一种改进的加性-显性(AD)模型进行eGWAS,该模型测试了每个标记的综合效应(加性效应和显性效应),以减少计算负担。在全基因组范围内,我们共鉴定出36,946个与18,517个e基因(与eQTL相关联的基因)相关的eQTL,占所有分析基因的64.51%,其中包括10,696个(28.95%)局部eQTL和26,250个(71.05%)远距离eQTL(图2A)。值得注意的是,局部eQTL分布在对角线上,而远距离eQTL则散布在整个基因组中(补充图11)。与非管家基因相比,管家基因(Lin等,2014)受到的eQTL调控显著较少(补充图12,Wilcoxon秩和检验的p值=8.29×10⁻⁶)。在这些e基因中,10,569个(57.08%)受到远距离eQTL调控,5,936个(32.06%)受到局部eQTL调控,2,012个(10.87%)同时受到局部和远距离eQTL调控(图2B)。在局部eQTL中,42.84%位于其目标e基因转录起始位点(TSS)的10kb范围内(图2C)。大多数e基因仅与一个eQTL相关联(图2D)。对eQTL中主要SNP的表型变异解释(PVE)值进行比较发现,局部eQTL对e基因表达的方差贡献大于远距离eQTL(图2E)。
为了鉴定与杂交性能相关的潜在调控节点和候选基因,我们分析了与QTL共定位的eQTL热点。通过对全基因组范围内的全局eQTL进行扫描,我们鉴定出552个eQTL热点,其中42个位于35个QTL的200kb范围内(补充图2F和补充表2)。例如,热点C10_Hot26与PH_Dom3(一个与株高相关的显性QTL)共定位,其中包含可能的候选基因GRF5(补充图13A)。C10_Hot26调控了69个e基因的表达(补充表3和图2G)。相关性分析显示,参与应激响应和生殖发育的e基因(如GSNAP、MNS1、WRKY11、CBR1、PPT1和ALY3)与GRF5表达呈正相关,但与株高(PH)呈负相关或无相关性。相比之下,参与营养代谢和信号转导的e基因(如SHR5、ATG8C和CML)与GRF5表达呈负相关,但与株高呈正相关(补充图13B)。对不同基因型杂交种在PH_Dom3位点的基因表达进行比较发现,携带纯合有利基因型(TT)的杂交种GRF5、CBR1和ALY3的表达降低,而ATG8C和CML的表达增加(补充图13C)。这些结果支持了这样一个假设:PH_Dom3的有利等位基因可能通过调节GRF5及其相关e基因的表达来增加株高。其他热点也得到了类似的结果。例如,热点C4_Hot21与PH_Dom18共定位,其中包含可能的候选基因SBP7,并与49个下游e基因相关,包括参与营养代谢和植物发育的关键基因(补充表3和图2G)。热点C7_Hot08与PH_Dom34共定位,并跨越了可能的候选基因CRF4。C7_Hot08与36个e基因相关,包括参与光合作用、营养代谢和细胞周期的关键基因(补充表3和图2G)。这些发现表明,这些QTL热点中的候选基因可能通过影响下游e基因的表达来调控株高。
共表达模块和模块eQTL(meQTL)的鉴定
为了进一步剖析杂交性能背后的调控网络,我们通过独立成分(IC)分析在杂交群体中鉴定出共表达模块。结果表明,共鉴定出205个独立成分为共表达模块(补充表4)。分配给IC模块的基因数量范围从10到841,中位数为250(补充图14)。IC的潜在特征与测试性状之间的相关性分析显示,25个IC与株高(PH)显著相关,47个IC与穗高(EH)显著相关,20个IC与这两个性状均显著相关(皮尔逊相关性,p < 0.01)(图3A和补充表4)。此外,与测试性状呈正相关的IC数量多于呈负相关的IC数量(图3A)。对与性状相关性最强的前10个正相关模块和前5个负相关模块进行基因本体(GO)富集分析发现,正相关模块中的基因在与植物营养生长相关的生物学过程中显著富集,而负相关模块中的基因在与植物生殖生长和应激响应相关的生物学过程中富集(补充表4;图3B和3C)。
为了识别在eGWAS中未被检测到的其他关键调控因子,我们使用AD模型对205个IC的潜在特征进行了模块eGWAS(meGWAS)。共鉴定出234个与84个IC相关的模块eQTL(meQTL),其中包括142个与46个性状相关IC相关的meQTL(图3D和补充表5)。我们采用了两种策略来验证这些meQTL的可靠性:首先,我们将这些meQTL的物理位置与eQTL热点的位置进行比较,发现125个(53.42%)meQTL位于eQTL热点的200kb范围内(补充表5)。其次,我们通过Fisher精确检验(见方法)测试了调控特定IC内基因的eQTL是否显著富集在相应meQTL的200kb侧翼区域,结果发现66个(28.21%)meQTL的侧翼区域存在显著富集(补充表5)。这些比较表明eGWAS和meGWAS结果之间高度一致,然而,meGWAS还检测到了一些不与任何eQTL热点共定位的meQTL。
接下来,我们将重点放在与性状相关IC模块相关的meQTL上。在与株高(PH)相关IC模块相关的104个meQTL中,28个(26.92%)与PH的QTL共定位,40个(38.46%)与eQTL热点重叠。此外,分配给相关IC模块的基因的eQTL在33个(31.73%)meQTL的侧翼区域表现出富集(图3E)。对于与穗高(EH)相关IC模块相关的101个meQTL,9个(8.91%)与EH的QTL共定位,45个(44.55%)与eQTL热点重叠,调控分配给相关IC模块基因的eQTL在37个(36.63%)meQTL的侧翼区域表现出富集(图3F)。基于分配给IC模块的基因构建的调控网络可能揭示了与meQTL共定位的QTL之间的调控关系。例如,PH_Add1(一个与株高相关的加性QTL)与一个调控IC187潜在特征的meQTL(IC187_C10M9366861)共定位,而IC187的6个基因受到同一区域的eQTL热点(C10_Hot10)的调控(补充图15A)。此外,IC187中的29个基因与GATA7(PH_Add1内的候选基因)表现出共表达(补充图15B)。分配给IC187的基因在碳水化合物代谢、微管组装和生长素生物合成等生物学过程中显著富集(图3B)。鉴于IC187与株高呈正相关,可以合理推测PH_Add1及其候选基因GATA7可能作为一个调控枢纽,通过上调模块IC187中的基因来控制株高。
类似地,与IC176潜在特征相关的meQTL(IC176_C8M170930342)与PH_Dom38(一个与株高相关的显性QTL)、EH_Dom6(一个与穗高相关的显性QTL)以及调控8个e基因的eQTL热点(C8_Hot40)共定位。该QTL包含了可能的候选基因MYB163(补充图16A)。在IC176的126个基因中,有4个受到C8_Hot40的调控(补充图16B)。基因本体(GO)富集分析显示,该模块中的基因在与植物生殖发育和应激响应相关的生物学过程中富集(图3C)。由于IC176与穗高呈负相关,可以合理推测PH_Dom38和EH_Dom6可能通过抑制模块IC176中的基因来调控株高和穗高。
bZIP29作为杂交生长的候选基因
为了选择一个用于功能分析的候选基因,我们将重点放在株高(PH)和穗高(EH)共有的三个显性QTL上:PH_Dom10(EH_Dom3)、PH_Dom38(EH_Dom6)和PH_Dom29(EH_Dom5)(补充表1),因为显性QTL对杂交性能变异的贡献大于加性QTL(补充图8-10)。我们发现PH_Dom10包含了bZIP29,而PH_Dom38包含了MYB163(补充表1)。值得注意的是,PH_Dom10与两个紧密连锁的eQTL热点(C1_Hot60和C1_Hot61;补充表2)共定位,当热点扫描距离阈值增加到>50kb时,这两个热点合并为一个单一热点(C1_Hot60*)(图4A)。此外,与IC181和IC176相关的4个meQTL也位于该区域(图3C和4A;补充表5)。因此,bZIP29被选为杂交生长的候选基因。
bZIP29编码一个基础亮氨酸拉链(bZIP)转录因子,并与QTL、eQTL和meQTL的峰值SNP表现出强连锁不平衡(LD)(图4B)。值得注意的是,bZIP29与这两个性状均显著正相关,且由C1_Hot60*调控的大多数目标e基因也与这两个性状和bZIP29表达显著相关(图4C)。在这些e基因中,AGD1(参与细胞骨架组织)、SLC35F5(编码溶质载体)、bHLH48(参与发育调控)、RPS17(30S核糖体蛋白)和PAP1(与叶绿体发育相关)与这两个性状和bZIP29表达呈正相关。相比之下,参与病害抗性或应激响应的基因(如GSTF8和UBP12)与bZIP29表达呈负相关(图4C)。bZIP29的局部eQTL可能驱动该基因在杂交群体中的差异调控,并影响下游e基因的表达。
一个具有显性效应的共同峰值SNP(C1M296443013)与这两个性状显著相关(图4A)。对C1M296443013不同基因型杂交种的杂交性能和MPH值进行比较发现,具有纯合有利基因型(CC)或杂合基因型(CT)的杂交种比具有纯合不利基因型(TT)的杂交种表现出更高的表型值和MPH值(图4D和4E;补充表6),这表明该位点对这两个性状及其MPH值具有显性效应。相比之下,具有杂合基因型的杂交种中bZIP29的表达水平显著低于具有纯合有利基因型的杂交种,但显著高于具有纯合不利基因型的杂交种(图4F),这表明bZIP29表达具有加性效应。值得注意的是,bZIP29表达与每个性状的MPH显著相关(补充图17),这表明bZIP29对株高和穗高均具有异质效应。此外,对三个基因型组的e基因表达水平进行比较发现,具有纯合有利或杂合基因型的杂交种比具有纯合不利基因型的杂交种表现出更低的负相关e基因(GSTF8和UBP12)表达水平(补充图18A)和更高的正相关e基因(SLC35F5、bHLH48和RPS17)表达水平(补充图18B)。这些结果表明,该QTL的有利等位基因可能通过影响bZIP29及其下游e基因的表达来增强株高和穗高。
为了进一步研究与bZIP29相关的功能变异,我们对峰值SNP(C1M296443013)和bZIP29基因体及其上游区域的所有潜在变异进行了连锁不平衡(LD)分析。两个变异——C1M296588268(一个SNP标记)和C1M296596845(一个插入/缺失标记)——与峰值SNP表现出强连锁不平衡(补充图19A)。值得注意的是,这两个变异都位于bZIP29的上游区域,表明它们可能作为顺式调控元件影响bZIP29的表达。
在围绕这两个变异的单倍型中,有四种基于单倍型的基因型(HapGs)在杂交群体中的频率超过0.05。随后对这些HapGs的bZIP29表型和表达水平进行比较发现,携带杂合单倍型(HapG2)的杂交种比携带纯合单倍型(HapG4)的杂交种表现出显著更高的株高(PH)、穗高(EH)和bZIP29表达水平(补充图19B)。这些发现突显了C1M296588268和C1M296596845作为验证bZIP29功能变异的强有力候选变异。
验证bZIP29对株高(PH)和穗高(EH)的异质效应
为了确认bZIP29的显性效应,我们获得了通过CRISPR-Cas9系统生成的两个独立的失功能突变体(DEL系)以及两个独立的过表达系(OE系)(Yang等,2022)。野生型(WT)系中bZIP29的表达水平显著高于DEL系及其衍生的F1代,而显著低于OE系及其衍生的F1代(图5A和5B),这反映了在杂交群体中观察到的加性表达模式(图4E)。DEL衍生的F1代植株比DEL系高,但比野生型矮(图5C和5D),表明对株高(PH)和穗高(EH)具有加性效应。然而,过表达bZIP29导致株高和穗高增加,在OE1F1中表现出超显性模式,而在OE2F1中表现出显性模式(图5E和5F)。两种OE衍生F1代之间的效应差异可能与bZIP29的表达水平有关。
我们对B73玉米自交系进行了两次高质量重复的tsCUT&Tag实验(图5G),以鉴定bZIP29蛋白靶向的下游基因。在两次生物学重复中,bZIP29蛋白共靶向了3363个基因。这些靶基因在多种生物学过程中显著富集,包括代谢、应激响应、发育和植物激素(补充图20)。值得注意的是,其中28个基因被21个QTL所包含(补充表7)。在由C1_Hot60*调控的18个e基因中,一个靶e基因(PTR3)的启动子被bZIP29蛋白结合。此外,IC176和IC181中的186个基因中有6个基因的启动子被bZIP29蛋白结合(图5H和5I;补充表7),表明在bZIP29蛋白结合的基因中e基因(卡方检验的p值=1.24×10⁻³)和模块基因(卡方检验p值=4.05×10⁻²⁷)显著富集。此外,其他e基因和模块基因可能受到bZIP29的间接调控。由于IC181中的基因富集于分生组织发育,而IC176中的基因富集于应激响应和减数分裂(图3C),bZIP29可能通过激活参与营养生长的基因并抑制与应激响应和生殖发育相关的基因来正向调控株高和穗高。通过整合e基因、两个IC中的基因以及bZIP29蛋白靶向的基因构建的复杂调控网络(图5J)表明,bZIP29参与了多个生物学过程。
以bZIP29为中心的调控网络在杂交种中比在亲本系中更为广泛和复杂
为了进一步研究bZIP29在杂种优势中的作用,我们将整个杂交群体根据四个父本测验系划分为四个亚群体,并比较了这些亚群体与亲本群体的以bZIP29为中心的共表达网络。通过皮尔逊相关性进行的共表达分析显示,与亲本群体相比,杂交亚群体中与bZIP29共表达的基因数量多出9.67到25.42倍(图6A)。此外,我们发现杂交群体和亲本群体中bZIP29的共表达网络既包含共同基因,也包含独特基因(图6A)。
基因本体(GO)富集分析显示,杂交亚群体中与bZIP29共表达的基因在与代谢过程、应激响应、发育过程和植物激素相关的常见GO术语中显著富集。相比之下,亲本群体中与bZIP29共表达的基因仅富集于四个GO术语(图6B)。此外,当相互比较时,杂交亚群体的共表达网络表现出相似的模式,并且比亲本群体的网络更为广泛和复杂(图6C)。这些发现表明,bZIP29通过参与在杂交亚群体中部分保守的复杂调控网络,对株高(PH)和穗高(EH)的杂种优势和杂交性能做出贡献。
讨论
系统遗传学方法已被用于鉴定玉米中与籽粒大小和苯并噁嗪类生物合成相关联的基因(Wang等,2018;Li等,2022)、棉花中的种子大小(Zhao等,2023)以及甘蓝型油菜中的开花时间和生长相关性状(Li等,2018)。在本研究中,我们首次将系统遗传学应用于杂交群体,以剖析玉米杂交性能的分子基础。我们对株高(PH)和穗高(EH)进行了全基因组关联研究(GWAS),并将其结果与表达GWAS(eGWAS)和模块eGWAS(meGWAS)相结合。基于QTL区域内注释基因的文献信息,我们鉴定出41%(33/79)的QTL候选基因。通过评估它们与eQTL和meQTL的共定位,我们优先考虑了六个QTL区域中的六个基因。我们评估了候选基因表达水平与测试性状之间的相关性,并分析了它们与位于eQTL热点和/或由meQTL调控的共表达模块(ICs)中的基因的共表达模式。这种系统遗传学方法适用于鉴定与杂交性能和杂种优势相关的候选基因,并为候选基因的分子网络提供了见解。值得注意的是,候选基因bZIP29位于一个影响株高和穗高的显性QTL中,并与一个eQTL热点和四个meQTL共定位,通过转基因实验被证实对测试性状具有异质效应。共表达分析显示,bZIP29在杂交亚群体中参与了比亲本群体更为广泛和复杂的网络。
选择株高(PH)和穗高(EH)研究玉米杂交性能和杂种优势的优势
为了研究玉米杂交性能和杂种优势的遗传基础,我们选择了两个具有高广义遗传力(H²)的性状,以减少环境因素的影响。鉴定出的79个QTL(补充表1)支持了这一选择。株高(PH)和穗高(EH)是强相关的性状(Li等,2017;Yin等,2022)(补充图4),因此我们认为这两个性状共有的QTL将是鉴定与杂交性能相关基因的特别有力的候选。鉴定出的候选基因MYB163和bZIP29分别位于四个共有QTL中的两个,证实了这一观点。尽管中亲优势(MPH)常被用于研究杂种优势,但它是一个基于性状表现计算得出的衍生复合性状,因此比杂交性能更容易受到意外因素的影响(图1B)(Li等,2021b)。因此,我们专注于杂交群体中的株高和穗高,而不是中亲优势。然而,鉴于杂交性能与中亲优势之间的高相关性(补充图5B和5C)(Li等,2021b),所获得的见解也可能对杂种优势研究具有相关性,例如跨越bZIP29的QTL。伸长节间的基因表达对于揭示株高和穗高的调控机制具有信息价值(Wang等,2022;Sun等,2024)。对多样化杂交群体的节间进行全基因组表达分析,揭示了与株高和穗高相关的多个基因调控网络,通过QTL与eQTL和meQTL的共定位,可以优先考虑与测试性状相关的候选基因。
多样化的NCII群体非常适合研究杂交性能和杂种优势的遗传基础
NCII杂交群体的亲本系表现出极大的遗传多样性,并且几乎没有群体结构,这使得候选基因的高分辨率定位成为可能。我们使用了整合加性亲缘关系矩阵和显性亲缘关系矩阵的加性-显性(AD)模型,通过分别测试标记的加性效应和显性效应来定位加性QTL和显性QTL。虽然在四个杂交亚群体中检测到的QTL效应并不一致,但在每个亚群体中都观察到了总体上的正向贡献(图1E和1F)。尽管不能完全排除假阳性结果,但QTL的可靠性得到了多方面的支持:显性QTL的显性程度大于加性QTL;所有检测到的QTL的累积效应与杂交性能表现出强相关性(图1E和1F);并且33个QTL包含与植物生长和发育相关的基因(补充表1)。对于株高(PH)和穗高(EH),显性QTL的效应大于加性QTL(补充图8A和8B;补充表1)。因此,我们的研究主要集中在显性QTL上,并优先对其中五个显性QTL的候选基因进行了详细分析。因此,与先前的报道一致(Xiao等,1995;Hashimoto等,2021;Li等,2021b),我们的结果支持显性QTL对杂交玉米杂交性能和杂种优势的重大贡献。
系统遗传学方法揭示了玉米杂交性能和杂种优势的候选基因
全基因组表达谱可用于定位单个基因或共表达网络模块(Feltus,2014)。通过整合GWAS、eGWAS和eQTL热点分析,我们发现35个QTL的200kb范围内有42个eQTL热点(补充表2)。我们详细研究了其中三个eQTL热点,并优先考虑了候选基因GRF5、SBP7和CRF4。选择这三个基因是因为它们与QTL和eQTL热点区域中的主要SNP表现出强连锁不平衡(LD)。此外,这三个基因均与植物发育或生长相关的基因表现出共表达(图2G)。由于编码序列中的突变可能会影响基因功能而不改变转录水平,因此那些与局部eQTL不相关的基因也被认为是候选基因。同样,全基因组阈值可能无法检测到某些局部eQTL。例如,GRF5基因在QTL的不同基因型类别之间表现出显著差异(补充图13),但未检测到局部eQTL。
GRF5、SBP7和CRF4的已报道功能与控制株高(PH)和穗高(EH)的假定作用一致。例如,含有SBP结构域的基因参与多种生长和发育过程。IPA1在水稻中调控植物株型(Song等,2017),TaSPL14在小麦中控制株高(Cao等,2021)。玉米中有30个SBP基因,这些基因被认为是改善高密度种植株型的有前途的候选基因(Wei等,2018)。GRF5已被证明在拟南芥中调控细胞大小、叶片大小和根长(Lantzouni等,2020),在杨树中调控叶片大小以及玉米素和异戊烯基腺嘌呤的含量(Wu等,2021a),并在一些双子叶和单子叶物种中调控转化效率(Kong等,2020)。CRF是AP2/ERF转录因子家族的一个小亚群基因,CRF4在拟南芥中调控生物量、根系发育和15NO₃吸收(Varala等,2018)。然而,需要注意的是,与候选基因连锁的基因也可能对eQTL热点有贡献。因此,对下游e基因的注释以及候选基因与下游e基因或测试性状之间的相关性分析(补充图13、15、16和图4C)对于为候选基因的功能角色提供辅助证据至关重要。
提取代表共表达基因组的关键特征可以降低维度,并有助于定位调控大量基因的顺式调控eQTL。我们认为模块eGWAS(meGWAS)可能会补充但也支持eGWAS的结果。事实上,在与性状相关模块相关的37个meQTL中,有13个(35.14%)位于QTL的10kb范围内,并且位于eQTL热点侧翼的10kb内(补充表5)。将meGWAS结果和共表达模块中的基因整合到分析中,即使相关的热点没有包含许多基因,也能为QTL区域提供详细信息,如跨越候选基因GATA7、MYB163和bZIP29的三个QTL所示。对其他植物物种中的候选基因GATA7和MYB163的研究支持它们在控制株高(PH)和穗高(EH)方面可能的功能。水稻中的OsGATA7调控油菜素内酯介导的植物生长和株型调控(Zhang等,2018),而MYB163属于R2R3型MYB转录因子,调控专门的代谢、发育和应激响应(Dubos等,2010)。
bZIP29是植物中一个新的异质基因
bZIP29被优先考虑为一个共同显性QTL的候选基因,该QTL与株高(PH)和穗高(EH)都相关,这是基于其与这两个性状以及相关热点和ICs中的下游基因的相关性(图4A-4C)。bZIP29上游区域的两个自然变异与bZIP29的表达显著相关,并可能对不同单倍型组之间的表型差异做出贡献(补充图19)。bZIP29的表达表现出很大的变异,并且与每个性状都有很强的正相关(图4C、4E和S17),这种相关性得到了过表达(OE)和缺失(DEL)系的支持。重要的是,bZIP29与杂交种群体中杂交性能和杂种优势的显性效应相关,转基因方法证实了其对株高和穗高的异质效应。
在拟南芥中,bZIP29调控植物生长和生殖器官的发育(Lozano-Sotomayor等,2016),其玉米同源基因通过与ZmABI19相互作用调控种子发育(Yang等,2022)。因此,bZIP29可能在植物发育的不同器官或不同阶段发挥作用。bZIP29的器官和阶段特异性表达支持了这一观点(www.maizegdb.org;MaizeNetome searchelement.php)。bZIP29蛋白的3363个靶基因在多种基因本体(GO)术语中富集的结果(补充图20)表明,bZIP29可能根据其器官和阶段特异性表达具有不同的功能角色。鉴于我们在本研究中验证了bZIP29在植物生长中的作用,并且其对粒重的影响已在先前的工作中得到证明(Yang等,2022),未来的研究值得探讨bZIP29对其他性状杂交性能的贡献。
除了影响杂种优势的大效应孟德尔位点外,也有证据表明上位性互作在杂种优势中发挥了重要作用(Jiang等,2017;Torgeman和Zamir,2023)。在多样化的杂交群体中研究上位性效应对于像本研究所使用的高密度SNP数据集来说在计算上具有挑战性。因此,我们使用了无法区分显性效应和上位性效应的加性-显性(AD)模型,有可能部分检测到的显性QTL实际上可能作为上位性QTL影响测试性状和基因表达水平的变异。考虑到bZIP29被一个显性QTL所跨越,并且参与了复杂的互作(图5J和补充表7),仅使用来自两个不同自交系的杂交系来验证其异质效应可能是有风险的。特别是,bZIP29蛋白与3363个基因发生互作,其中包括28个QTL跨越的基因(补充表7),并且作为18个e基因的核心转录调控因子(补充表2),其中一些与植物发育或逆境耐受性相关(图4C)。总之,推测bZIP29可能通过与其他基因的互作来调控杂交性能和杂种优势,这已在玉米的种子发育中得到证实(Yang等,2022)。
杂交系中既观察到加性表达,也观察到非加性表达,这些表达模式可能与杂种优势有关(Pea等,2008;Zhou等,2022;Zhang等,2023)。Springer和Stupar(2007)提出,在自交系中,基因表达水平可能过高或过低,从而对表型产生不利影响。杂交系中的中亲表达可能稀释这些不利影响,从而导致显性或超显性效应。过表达(OE)衍生的F1代是验证这一假设的理想材料。在本研究中,我们发现OE1系中的bZIP29表达水平高于OE2系,而OE1系中的不利影响也比OE2系更为严重,这表明bZIP29的表达水平与这些不利影响相关。在两种OE衍生的F1代中,bZIP29的表达水平均有所降低,从而缓解了bZIP29对株高(PH)的不利影响,导致这些性状在OE衍生的杂交系中表现出超显性和显性模式。这些结果与Springer和Stupar的假设一致。
在四个杂交亚群体中,bZIP29调控网络中的基因在共同的基因本体(GO)术语中表现出富集(图6B和6C),这表明具有这些GO术语的基因可能与杂交玉米的强健生长有关。值得注意的是,亲本群体中bZIP29的基因调控网络远小于杂交亚群体中的网络。在之前对代谢物相关网络进行分析的研究中也观察到了不同的网络特性,大多数玉米杂交种的网络密度高于相应的亲本系(Lisec等,2011)。同样,在拟南芥中,两个互交杂交种(Col0和C24)的连通性高于相应的亲本(Andorf等,2009)。基于这些结果,建议将关注互作的系统生物学方法视为对传统数量遗传学方法的补充,后者主要关注个体位点与遗传背景的上位性互作(Andorf等,2010)。未来专注于互作的研究应致力于破解杂交种中复杂调控网络的分子基础及其表型后果。
鉴定与杂交性能和杂种优势相关的基因已被证明是一项艰巨的任务;然而,一些成功的尝试为植物杂种优势的分子基础提供了极其宝贵的信息。OsMADS1是水稻中一个编码MIKC型MADS盒转录因子的基因,在F₂群体中表现出不完全显性。引入一个跨越OsMADS1内含子-外显子边界的15bp基因组片段改变了成熟mRNA的序列和翻译蛋白,从而导致粒长和粒重增加(Wang等,2019,2024)。通过整合遗传和转录组分析,推测RH8(也称为Ghd8)的等位基因杂合性是许多水稻杂交品种产量杂种优势的致因因素(Li等,2016)。在番茄中,开花基因SINGLE FLOWER TRUSS(SFT)在不同的遗传背景下对产量表现出超显性效应,其遗传效应与通过SELF PRUNING(SP)介导的生长终止的抑制相关(Krieger等,2010)。同样,BnaA9.CYP78A9是油菜中一个与产量杂种优势相关的异质基因。其上游调控区域的杂合性导致在表达水平和生长素水平上表现出部分显性,从而导致下游基因的非加性表达(Ye等,2023)。基于拟南芥半双列杂交群体的GWAS分析显示,AGL50(AGAMOUS-LIKE 50)显性控制开花时间,并在四种遗传背景下验证了AGL50的功能(Seymour等,2016)。在玉米中,群体分析显示ZAR1和ZmACO2对产量性状表现出超显性效应(Wang等,2023)。我们的发现与先前的结果一起表明,植物中的杂种优势与许多不同的途径有关,尽管取得了这些进展,但关于作物植物中杂种优势如何调控的信息仍然支离破碎。
总之,本研究描述了玉米杂交群体中QTL的调控关系。通过结合GWAS、eGWAS、meGWAS和Fisher精确检验,优先鉴定了六个潜在的异质基因,其中一个基因bZIP29被一个影响株高(PH)和穗高(EH)的显性QTL、两个eQTL热点以及两个ICs中的四个meQTL所跨越。跨越bZIP29的QTL对杂交性能和杂种优势表现出显性效应。转基因实验证实了bZIP29对植物生长表现出异质效应。tsCUT&Tag实验和调控网络分析揭示了涉及bZIP29的杂种优势的调控机制。本研究揭示了影响玉米杂交性能和杂种优势的关键基因和调控网络。
方法
群体构建、表型评估和表型数据分析
从包含527个具有热带、亚热带和温带背景的自交系的大型玉米关联作图小组中选择了78个母本自交系(补充表8)(Yang等,2011;Li等,2013a;Gui等,2022)。我们从关联作图小组的温带亚群中选择了78个母本系,以便获得的杂交系能够在中国北方正常生长和开花。所选母本系的系统发育分布相对平衡,没有极端分化的类群(补充图2A),这有助于在多样化的杂交群体中对杂交性状进行遗传作图。
所用的四个父本系M01、M02、M03和M04在我们之前的研究中使用过(Li等,2021a),并且它们属于不同的类群(补充图2A)。按照北卡罗来纳II(NCII)设计构建了杂交群体,产生了四个杂交亚群体,每个亚群体包含78个杂交系。通过将M01与M02杂交以及将M03与M04杂交,获得了两个杂交系。总共使用了314个杂交系进行表型调查和转录组测序。
2018年夏季,杂交群体在涿州(ZZ;河北省,北纬39°29’,东经115°58’)、新乡(XX;河南省,北纬35°22’,东经113°54’)和公主岭(GZL;吉林省,北纬43°30’,东经124°49’)播种。每个杂交系以两行种植,行距为60厘米,株距为25厘米。群体采用不完全区组设计,每个区组包含一个杂交亚群体。在开花后阶段,对每个小区的10株植株的株高(PH)和穗高(EH)进行评估,并排除超出1.5倍四分位距的异常值。
最佳线性无偏估计(BLUEs)的计算分为两个步骤,如先前研究所述(Jiang等,2017)。简而言之,在第一步中,通过包含基因型、重复和嵌套在重复中的区组效应的线性混合模型,获得每个环境下群体的BLUEs。基因型效应作为固定效应,其他效应作为随机效应。在第二步中,通过将基因型和环境效应分别作为固定效应和随机效应的线性混合模型,获得跨环境的BLUEs。父母本和杂交系的BLUEs用于后续的遗传分析。中亲优势(MPH)按以下公式计算:
MPH = BLUE F 1 − ( BLUE M + BLUE F ) 2 \text{MPH} = \frac{\text{BLUE}_{F1} - \left( \text{BLUE}_M + \text{BLUE}_F \right)}{2} MPH=2BLUEF1−(BLUEM+BLUEF)
为了估计广义遗传力(H²),通过拟合一个将所有效应均视为随机效应的线性混合模型来计算最佳线性无偏预测(BLUPs)。H²按以下公式计算(Cullis等,2006):
H 2 = 1 − v BLUP 2 σ G 2 H^2 = 1 - \frac{v_{\text{BLUP}}}{2\sigma^2_G} H2=1−2σG2vBLUP
其中,( σ G 2 \sigma^2_G σG2)是基因型方差,( v BLUP v_{\text{BLUP}} vBLUP) 是每对BLUPs差异的平均方差。
转录组测序与分析
2018年6月,所有314个杂交系在北京海淀区播种。当植株达到V7阶段时,每行选取三株植株的去顶幼嫩节间混合为一个样本。使用RNeasy植物小柱试剂盒(Qiagen,德国)提取总RNA,并使用凯奥K5500分光光度计(凯奥,北京,中国)检测RNA纯度。使用Bioanalyzer 2100系统(Agilent Technologies,美国加州圣克拉拉)的RNA Nano 6000检测试剂盒评估RNA完整性和浓度。通过连接多聚T寡核苷酸的磁珠从总RNA中纯化mRNA。随后使用NEBNext Ultra Directional RNA Library Prep Kit for Illumina(NEB,美国马萨诸塞州Ipswich)构建测序文库,并在Illumina NovaSeq平台上进行测序,获得150bp的双端测序数据。每个文库获得了超过10Gb的数据。
为了定量分析基因表达,首先使用Trimmomatic-0.36(Bolger等,2014)去除接头序列、低质量碱基(质量评分<20)和短读长(长度<30bp)的原始RNA-seq数据进行过滤。然后使用STAR(v.2.6)以双通道模式(Dobin等,2013)将过滤后的读段比对到玉米参考基因组(ARGv4),并提取唯一比对的读段用于基因表达定量。使用HTseq-count(参数:-s reverse -mode union)(Anders等,2015)计算玉米B73 ARGv4基因组中注释基因的读段计数。使用DESeq2(Love等,2014)校正文库大小,并计算基因的每千碱基每百万比对片段数(FPKM)的标准化值。通过过滤杂交群体中FPKM值的中位数>0的基因,获得了28,706个过滤后的基因,并使用R语言的qqnorm()函数对其表达水平进行标准化。使用PEER(Stegle等,2010)估计表达谱中的技术变异,检测到16个隐藏的混杂因子。
利用全基因组重测序数据进行SNP鉴定
使用公开的亲本系全基因组重测序数据(Gui等,2022)进行SNP鉴定。首先,使用Trimmomatic-0.36(Bolger等,2014)去除接头序列、低质量碱基(质量评分<20)和短读长(长度<30bp)对原始读段进行过滤。然后,使用BWA-MEM(Li,2013)以默认参数将处理后的读段比对到玉米参考基因组(ARGv4)。仅提取唯一比对的读段用于SNP鉴定,该过程遵循GATK(v.3.8.1)变异鉴定的最佳实践指南(DePristo等,2011)。简而言之,使用Picard Tools(Picard Tools - By Broad Institute)对比对文件(BAM格式)进行排序、标记重复读段、添加分组信息并进行索引。接下来,使用GATK的IndelRealigner对包含插入/缺失(indels)的读段进行重新比对,并使用GATK的BaseRecalibrator进行碱基校准,以玉米HapMap v.3.2.1中的SNPs作为已知位点(Bukowski等,2018)。使用HaplotypeCaller进行基因组变异鉴定,参数设置为:-dontUseSoftClippedBases -stand_call_conf 20.0 -ERC GVCF -variant_index_type LINEAR。使用GenotypeGVCFs对所有自交系进行联合基因分型。最后,使用质量参数(QD≥2.0、FS≤60.0、MQ≥20.0)对原始SNPs进行过滤。
为了准备用于遗传分析的SNP标记,通过质量控制的SNPs进一步过滤,要求亲本群体中的杂合率≤20%且缺失率≤20%。使用Beagle(Browning等,2018)对过滤后的SNPs进行填充,主要参数设置为window=50000和overlap=10000。亲本群体的加性基因型编码为0、1和2,其中0表示主要纯合基因型,1表示杂合基因型,2表示次要纯合基因型。由于亲本群体样本量有限,SNP位点之间存在广泛的长距离连锁不平衡(LD)。为了去除这些SNPs,我们采用了Seymour等(2016)提出的方法。简而言之,在加性基因型矩阵中,我们将全群体具有相同基因型的SNPs视为一种基因型组合,发现共有1,823,526种基因型组合,这些组合中的SNPs数量从1到33,586不等。我们保留了包含少于50个SNPs的组合及其对应的SNPs。此外,还去除了在染色体上表现出完全长距离LD的SNPs。最终,亲本群体获得了4,628,240个SNPs。杂交系的基因型是根据其亲本的基因型推断而来的。
基于基因型数据计算的距离矩阵构建了邻接(Neighbor-Joining, NJ)树。NJ树的构建使用了R语言的APE包(Paradis等,2004)。
杂交性能的GWAS分析和QTL鉴定
采用包含加性效应和显性效应的线性混合模型(AD模型)进行全基因组关联研究(GWAS)。AD模型(Bonnafous等,2018;Ramstein等,2020)表示为:
[ y = m + x i q a i + z i q d i + A + D + e y = m + x_i q_{a_i} + z_i q_{d_i} + A + D + e y=m+xiqai+ziqdi+A+D+e ]
(公式3)
其中,( y ) 是最佳线性无偏估计(BLUEs);( m ) 是截距;( x i x_i xi ) 和 ( z i z_i zi ) 分别是第 ( i ) 个SNP的加性基因型和显性基因型;( q a i q_{a_i} qai ) 和 ( q d i q_{d_i} qdi) 分别是第 ( i ) 个SNP的加性效应和显性效应;( A ) 是随机加性效应;( D ) 是随机显性效应。假设 ( A ∼ N ( 0 , σ a 2 K a ) A \sim N(0, \sigma_a^2 K_a) A∼N(0,σa2Ka) ),( D ∼ N ( 0 , σ d 2 K d ) D \sim N(0, \sigma_d^2 K_d) D∼N(0,σd2Kd) ),其中 ( K a K_a Ka ) 是加性亲缘关系矩阵(VanRaden,2008),( K d K_d Kd ) 是显性亲缘关系矩阵(Vitezica等,2013)。加性基因型的编码方式如前文所述。对于显性基因型矩阵,每个SNP的纯合基因型和杂合基因型分别编码为0和1。为了降低计算负担,基于预先确定的群体参数(P3D)算法(Zhang等,2010),将混合模型近似转换为简单的线性回归模型。通过t检验确定每个标记的加性效应和显性效应的显著性。在关联作图中使用了2,324,652个SNP,这些SNP的每种基因型(两种纯合基因型和一种杂合基因型)的频率均大于0.05。
基于有效标记的数量(Me)确定全基因组关联分析的显著性阈值。通过GEC(v.0.2)(Li等,2012)鉴定了74,316个有效SNP。为了确定最佳阈值,比较了四种方法:(1)根据先前的建议(Li等,2013a;Wen等,2014),采用1/Me确定的修正Bonferroni校正阈值为1.34×10⁻⁵;(2)计算为0.05/Me的Bonferroni校正阈值为6.73×10⁻⁷;(3)使用R语言中的p.adjust()函数实现的假发现率(FDR)阈值,FDR < 0.05;(4)置换检验(PT)。对于PT阈值,将每个性状的BLUE值随机置换1000次,并使用AD模型进行GWAS。将对应于单尾概率为0.01的p值设定为经验阈值。比较了这四种方法,观察结果如下(补充表9):第二种方法对于EH没有鉴定出显性QTL,对于两个性状的加性QTL也仅有少数;第三种方法对于PH没有鉴定出加性QTL,对于EH没有鉴定出显性QTL;第四种方法鉴定出的QTL超过200个。第一种方法更好地反映了株高(PH)和穗高(EH)的数量遗传模式,因此采用第一种方法确定QTL显著性阈值。
杂交性能的QTL鉴定步骤
杂交性能的QTL鉴定采用以下步骤进行。首先,提取所有具有加性效应和显性效应的显著SNP,并以200kb的距离阈值将它们聚类成区间。接下来,仅保留跨越至少3个显著SNP的区间,并将其视为QTL。区间/QTL中的主要SNP被用来代表该QTL。使用基于似然比的R²来估计主要SNP解释的表型变异(PVE),该模型包含加性亲缘关系矩阵和显性亲缘关系矩阵(Sun等,2010)。模型拟合使用了GridLMM包(Runcie和Crawford,2019)。显性效应与加性效应的比值(d/a)被用来表示每个QTL的显性程度。加性效应(a)是两种纯合基因型平均表型值之差的一半。显性效应(d)是杂合基因型与中亲值之间的表型差异。每个QTL的主要SNP的有利等位基因和不利等位基因定义如下:携带纯合有利等位基因的杂交种在杂交群体中应表现出比携带纯合不利等位基因的杂交种更高的平均表型值。
每个杂交种的“有利”评分是通过计算所有检测到的QTL中主要SNP的数值基因型之和得出的。简而言之,首先根据遗传效应将每个QTL中主要SNP的基因型编码为数值格式。对于加性QTL,主要SNP的基因型被编码为1、0或-1,其中1表示纯合有利基因型,0表示杂合基因型,-1表示纯合不利基因型。对于显性QTL,主要SNP的基因型被编码为1和0,其中1表示杂合基因型,0表示纯合基因型。因此,“有利”评分表示每个杂交种中所有检测到的QTL的有利基因型的累积效应。
eQTL和热点分析
eGWAS分析采用修改自公式3的AD模型进行。在此模型中,将16个PEER因子作为固定效应纳入,以校正技术变异,并通过Wald检验测试每个标记的综合效应(加性效应和显性效应)的显著性。为了降低eQTL定位的计算负担,使用PLINK进行基于全基因组连锁不平衡(LD)的筛选,去除冗余SNP((r^2 \geq 0.99)),关键参数设置为--indep-pairwise 100 5 0.99,最终得到889,673个具有信息量的SNP。显著性阈值(1.34×10⁻⁵)与GWAS分析一致。
对于特定基因,eQTL的鉴定步骤如下:(1)将距离在10kb范围内的显著SNP(eSNP)归为一个候选eQTL区间,并去除包含少于3个eSNP的候选eQTL。选择主要SNP来代表候选eQTL。(2)如果多个候选eQTL之间表现出强连锁不平衡((r^2 > 0.1)),则去除显著性较低的eQTL。eQTL的目标基因被称为e基因。根据eQTL与目标基因之间的相对距离(以200kb为阈值),确定局部eQTL和远距离eQTL。使用hot_scan软件(Silva等,2014)通过设置窗口大小为10kb和调整后p值的阈值为<0.01来鉴定eQTL热点。
IC模块和meQTL鉴定
独立成分(IC)分析使用picaplot软件包(版本0.99.7)(GitHub - jinhyunju/picaplot)进行,采用fastICA算法,关键参数设置为n_runs=20、n_cores=40、max_iter=500,以鉴定共表达模块。使用95%累积方差的截断值确定最优的IC数量。使用R软件包fdrtool将基因分配到每个模块中。对于源信号权重的FDR估计,调整后的p值小于0.001的基因被添加到模块中。最后,仅保留分配了10个或更多基因的IC。
对IC模块的潜在特征进行meGWAS分析,使用与eGWAS分析中描述的相同的AD模型。采用与鉴定eQTL相同的流程来鉴定meQTL。为了检查eQTL是否在meQTL的200kb侧翼区域内显著富集,进行了Fisher精确检验(Sun等,2023),分为以下四类:(1)分配给特定IC模块的e基因,这些基因受到调控该特定IC模块的meQTL侧翼区域内的eQTL调控;(2)未分配给特定IC模块的e基因,这些基因受到调控该特定IC模块的meQTL侧翼区域内的eQTL调控;(3)分配给IC模块但未受到调控该特定IC模块的meQTL侧翼区域内的eQTL调控的基因;(4)既未分配给IC模块也未受到调控该特定IC模块的meQTL侧翼区域内的eQTL调控的e基因。显著的p值(<0.05)表明,与偶然预期相比,分配给IC模块的基因中有更多基因的eQTL与meQTL相关联。
使用玉米-GAMER注释(Wimalanathan等,2018)对每个IC模块中分配的基因进行基因本体(GO)功能富集分析,并使用GOATOOLS(Klopfenstein等,2018)通过超几何检验(Bonferroni校正后的p值<0.05)实现。
bZIP29的单倍型分析
基于重测序的变异(使用GATK鉴定的SNPs和indels)位于bZIP29基因体及其上游10kb区域内,根据质量和基因型频率(杂交群体中三种基因型的频率>0.05)进行过滤。这些变异随后用于株高(PH)和穗高(EH)的关联分析,采用AD模型。显著性与性状相关的变异(p < 0.01)被聚类成基因型组,使用R语言中的单因素方差分析模型(aov(trait ~ group, data))进行分析。最显著的基因型组被视为相关联的单倍型。通过Tukey的HSD检验(调整后的p < 0.05)对均值进行成对比较,进一步评估相关联单倍型的表型和表达水平效应;只有频率超过0.05的单倍型组(HapGs)被包括在成对比较中。
转基因系的构建与表型调查以及通过RT-qPCR定量bZIP29的表达
bZIP29 CRISPR-Cas9转基因系(DEL)和bZIP29过表达转基因系(OE)的构建在之前的研究中已有描述(Yang等,2022)。DEL和OE系被可靠地用于比较转基因系与野生型(WT)之间的种子发育差异。DEL系是通过农杆菌介导的转化在B104自交系背景下生成的。得到的DEL植株被杂交并回交到B104中,以选择不含Cas9构建体的BC1代种子。选定的BC1植株自交以获得野生型和纯合DEL系。bZIP29的全长编码序列(CDS)由27-kDa γ-zein启动子驱动,克隆到pTF102质粒中,转化到Hi-II杂交系中,然后回交到B104超过四代。OE系自交后代中的空位分离体被用作野生型对照。
2024年夏季在北京对转基因系的表型进行了调查,测量了DEL、OE、WT及其F1代的株高和穗高。在V7阶段收集DEL、OE、WT及其F1代的幼嫩节间,用于定量bZIP29的表达。使用FastPure Universal Plant Total RNA Isolation Kit(货号RC411;南京Vazyme)提取总RNA。对于每个样本,使用1ng RNA通过HiScript II Q RT SuperMix for qPCR(+gDNA wiper)(Vazyme)合成第一条链的互补DNA(cDNA)。RT-qPCR使用TaqPro Universal SYBR qPCR Master Mix(Vazyme)在CFX-96实时PCR平台上(Bio-Rad,美国加州赫拉克勒斯)进行。以ZmActin基因为内参(Zhang等,2020)。用于RT-qPCR的引物序列列于补充表10。
tsCUT&Tag实验及数据分析
tsCUT&Tag技术被用于分析bZIP29的调控网络(Wu等,2022)。实验操作使用了针对Illumina平台的Hyperactive In-Situ ChIP Library Prep Kit(pG-Tn5)(Vazyme TD901)。通过荧光显微镜观察B73的转化原生质体以检测转化效率。选择转化效率大于60%的样本用于后续的tsCUT&Tag实验。为包含bZIP29 CDS的构建载体设置了两个生物学重复。通过低速离心(100转/分钟,持续2分钟)收集细胞,并将其重悬于100 mL重悬液中。在用刀豆蛋白A(Concanavalin A)珠子处理后,样本与GFP抗体及其对应的二抗一起孵育。使用pG-Tn5转座酶对DNA进行片段化并插入接头。最后,提取片段化的DNA用于文库构建。使用Qubit进行定量后,文库在Illumina HiSeq X-Ten平台上进行测序,以获得150bp的双端测序数据。转化了pM999-GFP载体的原生质体被用作对照。使用Bowtie 2(Langmead和Salzberg,2012)将清理后的读段比对到B73参考基因组(AGPv4),并使用Macs(Feng等,2012)在全基因组范围内扫描高置信度峰(ppeak < 0.0001)。使用R语言中的ChIPseeker(Yu等,2015)分析全基因组范围内峰的分布。如果一个峰位于基因转录起始位点(TSS)的3kb范围内,则该基因被认为是目标基因。
数据和代码的可用性
玉米杂交种的RNA-seq数据已存储于NCBI SRA数据库,访问编号为NCBI:PRJNA1000225。玉米亲本系的全基因组重测序数据从NCBI SRA数据库下载,访问编号为NCBI:PRJNA531553。