R包安装报错解决案例系列|R包使用及ARM架构解决data.table安装错误问题
有不少同学是Mac系统的,分析过程中会发现部分R包总是安装不成功,这是因为部分R包基于windowsx86架构编译的,最常见的就是含 C/C++/Fortran 的包,对于初学者都是建议linux和win去做,Windows 通常直接安装预编译好的二进制包。mac这种半开源的封闭性系统会很折腾,除非重新编译嘿嘿嘿,能把人折磨麻了!!!!!
绝大部分都是因为Apple不支持openmp等这类,需要到多线程加速运行的R包,必翻车。没办法,喜欢 Mac OS 得折腾!!!
文章目录
- data.table报错的出现
- 情况1 源代码编译
- 情况2 内置C++加速的并行OpenMP
- 情况3 退而求其次
- 情况4 安装中间R包
- 还有什么R包安装疑难杂症!!留言!!!
- 我也不信邪了,得折腾!!!!
data.table报错的出现
这个问题非常常见,这里举例两个这几天多人问到的,TCGAbiolinks和maftools都需要的一个依赖包 data.table
Error: package or namespace load failed for ‘data.table’ in dyn.load(file, DLLpath = DLLpath, ...):unable to load shared object '/Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/library/00LOCK-data.table/00new/data.table/libs/data_table.so':dlopen(/Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/library/00LOCK-data.table/00new/data.table/libs/data_table.so, 0x0006): symbol not found in flat namespace '___kmpc_dispatch_deinit'
Error: loading failed
Execution halted
ERROR: loading failed
* removing ‘/Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/library/data.table’
* restoring previous ‘/Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/library/data.table’The downloaded source packages are in‘/private/var/folders/r1/fyhb81050xd7jv1g6r1783dr0000gn/T/RtmptW7Nf8/downloaded_packages’
Warning message:
In install.packages(update[instlib == l, "Package"], l, repos = repos, :installation of package ‘data.table’ had non-zero exit status
我的Mac版本是macOS Sequoia 15.5,这是测试电脑的R环境:
> sessionInfo()
R version 4.4.3 (2025-02-28)
Platform: aarch64-apple-darwin20
Running under: macOS Sequoia 15.5Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8time zone: Asia/Shanghai
tzcode source: internalattached base packages:
[1] stats graphics grDevices utils datasets methods base other attached packages:
[1] ggplot2_3.5.2 TCGAbiolinks_2.34.1 maftools_2.22.0 BiocParallel_1.40.2 data.table_1.17.2 tibble_3.2.1 stringr_1.5.1
[8] dplyr_1.1.4 loaded via a namespace (and not attached):[1] tidyselect_1.2.1 farver_2.1.2 blob_1.2.4 R.utils_2.13.0 filelock_1.0.3 [6] Biostrings_2.74.1 fastmap_1.2.0 BiocFileCache_2.14.0 promises_1.3.2 XML_3.99-0.18
[11] digest_0.6.37 lifecycle_1.0.4 processx_3.8.6 survival_3.8-3 KEGGREST_1.46.0
[16] RSQLite_2.3.11 magrittr_2.0.3 compiler_4.4.3 rlang_1.1.6 progress_1.2.3
[21] tools_4.4.3 knitr_1.50 prettyunits_1.2.0 S4Arrays_1.6.0 bit_4.6.0
[26] curl_6.2.3 DelayedArray_0.32.0 plyr_1.8.9 xml2_1.3.8 RColorBrewer_1.1-3
[31] websocket_1.4.4 abind_1.4-8 purrr_1.0.4 withr_3.0.2 R.oo_1.27.1
[36] BiocGenerics_0.52.0 grid_4.4.3 stats4_4.4.3 scales_1.4.0 biomaRt_2.62.1
[41] SummarizedExperiment_1.36.0 cli_3.6.5 crayon_1.5.3 generics_0.1.4 remotes_2.5.0
[46] rstudioapi_0.17.1 httr_1.4.7 tzdb_0.5.0 chromote_0.5.1 DBI_1.2.3
[51] DNAcopy_1.80.0 cachem_1.1.0 zlibbioc_1.52.0 splines_4.4.3 rvest_1.0.4
[56] parallel_4.4.3 AnnotationDbi_1.68.0 TCGAbiolinksGUI.data_1.26.0 BiocManager_1.30.25 XVector_0.46.0
[61] matrixStats_1.5.0 vctrs_0.6.5 Matrix_1.7-3 jsonlite_2.0.0 IRanges_2.40.1
[66] hms_1.1.3 S4Vectors_0.44.0 bit64_4.6.0-1 tidyr_1.3.1 glue_1.8.0
[71] ps_1.9.1 codetools_0.2-20 stringi_1.8.7 gtable_0.3.6 later_1.4.2
[76] GenomeInfoDb_1.42.3 GenomicRanges_1.58.0 UCSC.utils_1.2.0 pillar_1.10.2 rappdirs_0.3.3
[81] GenomeInfoDbData_1.2.13 R6_2.6.1 dbplyr_2.5.0 httr2_1.1.2 vroom_1.6.5
[86] evaluate_1.0.3 lattice_0.22-7 Biobase_2.66.0 readr_2.1.5 R.methodsS3_1.8.2
[91] png_0.1-8 memoise_2.0.1 Rcpp_1.0.14 SparseArray_1.6.2 xfun_0.52
[96] downloader_0.4.1 MatrixGenerics_1.18.1 pkgconfig_2.0.3
为什么会这样,猜测
- data.table 包用到了 OpenMP 并行支持(C/C++层面),系统在编译时找不到特定的 OpenMP 符号(如 ___kmpc_dispatch_deinit)
- 在 macOS 上 R 是通过 Homebrew、M1 架构下的 clang 编译,OpenMP 配置不完整
不同人会有不同情况的,真的,有的提前独立安装好依赖包后直接重启一下R环境又醒了,有的却要重新编译。不同情况大家借鉴一下,绕不开的,都是libomp、libxm12、openssl这些系统库,或者像gfortran,或者环境变量,或者一些中间R包没安装成功。。。出现情况都谷歌一下,老外的论坛里很多踩过的坑比我们都早个1-2年,兴许都有解决方案
情况1 源代码编译
这是直接去源代码编译的,
https://stackoverflow.com/questions/65251887/clang-7-error-linker-command-failed-with-exit-code-1-for-macos-big-sur/65334247?answertab=modifieddesc#tab-top

如果还失败了考虑是不是这种情况,重新安装gfortran,https://stackoverflow.com/questions/65860439/installing-data-table-on-macos

情况2 内置C++加速的并行OpenMP
针对于部分机器学习的R包会出现,例如xgboost、lightgbm这类强依赖的,在 macOS 系统中,默认编译器不支持 OpenMP,需额外安装 libomp 并配置编译参数,常成为某些 R 包在 Apple M 系列芯片上安装失败的根源。
https://mac.r-project.org/openmp/
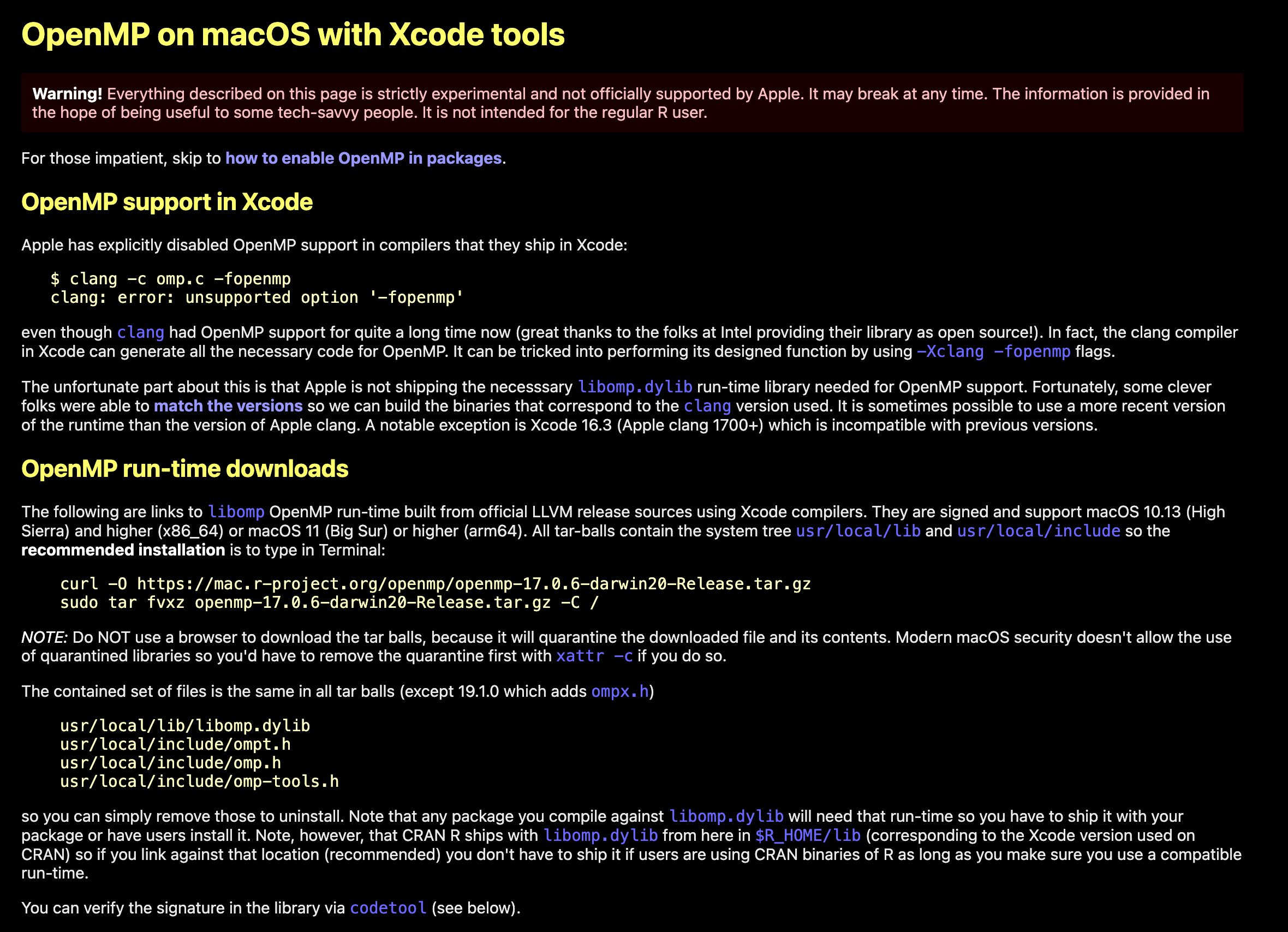
还有一个网址,作者的教程也很详细
https://investcookies.ru/post/datatable_m1/data_table_arm/
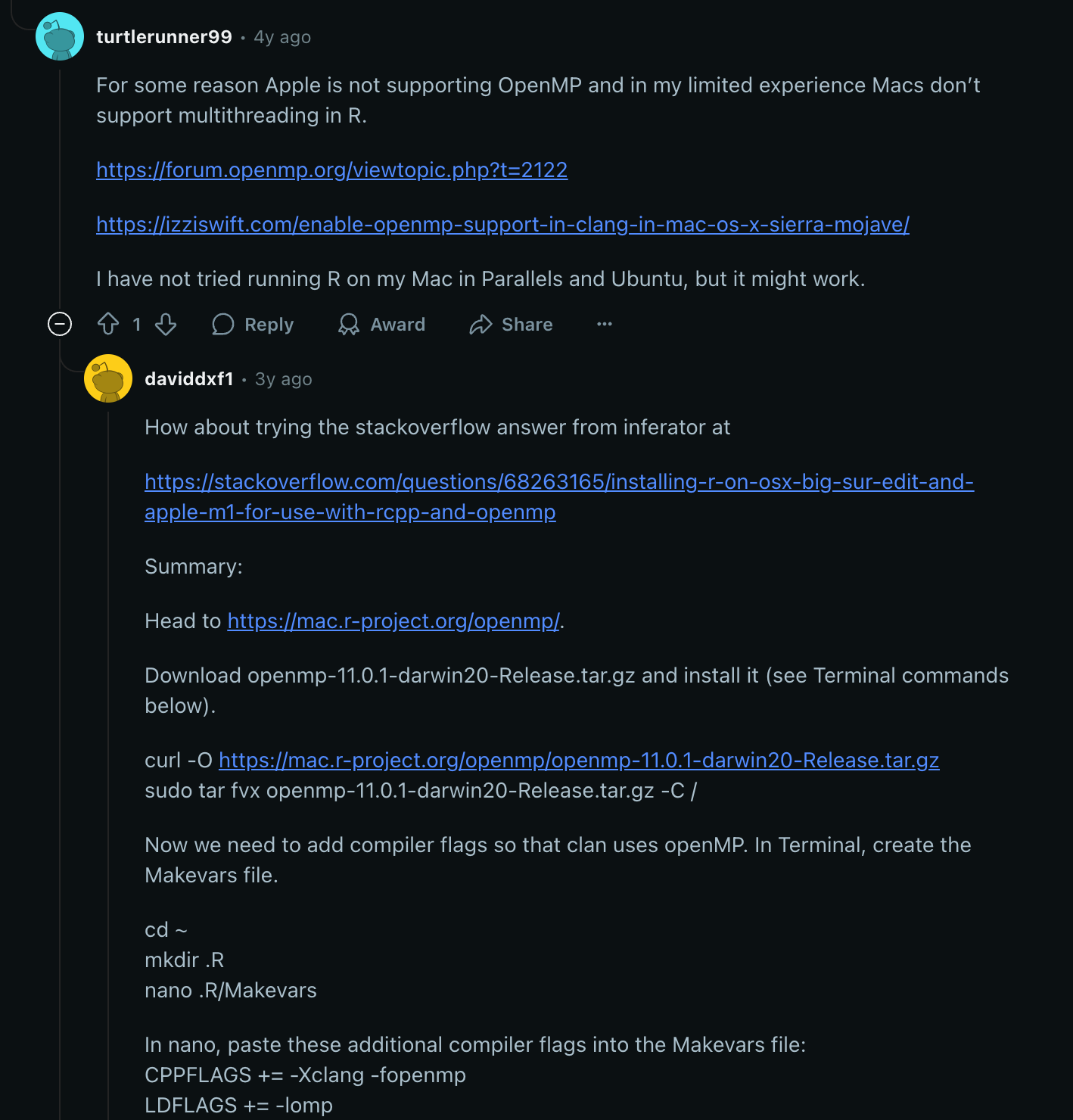
情况3 退而求其次
本质上还是因为这些R包采用的多线程,但是很多时候没有考虑到M芯片的使用环境,怎么办呢?换,换成像基于 dplyr 这种单线程的包,或者换 win/linux 吧。
情况4 安装中间R包
恭喜你,最简单的一种方式,去看那个报错,看具体是什么原因,有r包没装成功,独立去下载安装包,本地安装!!
> BiocManager::install("maftools",force=T)
'getOption("repos")' replaces Bioconductor standard repositories, see '?repositories' for detailsreplacement repositories:CRAN: https://cran.rstudio.com/Bioconductor version 3.16 (BiocManager 1.30.19), R 4.2.2 (2022-10-31)
Installing package(s) 'maftools'
trying URL 'https://bioconductor.org/packages/3.16/bioc/bin/macosx/contrib/4.2/maftools_2.14.0.tgz'
Content type 'application/x-gzip' length 11567249 bytes (11.0 MB)
==================================================
downloaded 11.0 MBThe downloaded binary packages are in/var/folders/bx/n5y8wbvx6td4rk2q0ht795lm0000gn/T//Rtmp8t6yAm/downloaded_packages> library("maftools")
Error: package or namespace load failed for ‘maftools’ in dyn.load(file, DLLpath = DLLpath, ...):unable to load shared object '/Library/Frameworks/R.framework/Versions/4.2/Resources/library/DNAcopy/libs/DNAcopy.so':dlopen(/Library/Frameworks/R.framework/Versions/4.2/Resources/library/DNAcopy/libs/DNAcopy.so, 0x0006): Symbol not found: (__gfortran_os_error_at)Referenced from: '/Library/Frameworks/R.framework/Versions/4.2/Resources/library/DNAcopy/libs/DNAcopy.so'Expected in: '/Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libgfortran.5.dylib'
> BiocManager::install("DNAcopy",force=T)
'getOption("repos")' replaces Bioconductor standard repositories, see '?repositories' for detailsreplacement repositories:CRAN: https://cran.rstudio.com/Bioconductor version 3.16 (BiocManager 1.30.19), R 4.2.2 (2022-10-31)
Installing package(s) 'DNAcopy'
trying URL 'https://bioconductor.org/packages/3.16/bioc/bin/macosx/contrib/4.2/DNAcopy_1.72.1.tgz'
Content type 'application/x-gzip' length 510684 bytes (498 KB)
==================================================
downloaded 498 KBThe downloaded binary packages are in/var/folders/bx/n5y8wbvx6td4rk2q0ht795lm0000gn/T//Rtmp8t6yAm/downloaded_packages> BiocManager::install("maftools",force=T)
'getOption("repos")' replaces Bioconductor standard repositories, see '?repositories' for detailsreplacement repositories:CRAN: https://cran.rstudio.com/Bioconductor version 3.16 (BiocManager 1.30.19), R 4.2.2 (2022-10-31)
Installing package(s) 'maftools'
trying URL 'https://bioconductor.org/packages/3.16/bioc/bin/macosx/contrib/4.2/maftools_2.14.0.tgz'
Content type 'application/x-gzip' length 11567249 bytes (11.0 MB)
==================================================
downloaded 11.0 MBThe downloaded binary packages are in/var/folders/bx/n5y8wbvx6td4rk2q0ht795lm0000gn/T//Rtmp8t6yAm/downloaded_packages> library("maftools")
Error: package or namespace load failed for ‘maftools’ in dyn.load(file, DLLpath = DLLpath, ...):unable to load shared object '/Library/Frameworks/R.framework/Versions/4.2/Resources/library/DNAcopy/libs/DNAcopy.so':dlopen(/Library/Frameworks/R.framework/Versions/4.2/Resources/library/DNAcopy/libs/DNAcopy.so, 0x0006): Symbol not found: (__gfortran_os_error_at)Referenced from: '/Library/Frameworks/R.framework/Versions/4.2/Resources/library/DNAcopy/libs/DNAcopy.so'Expected in: '/Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libgfortran.5.dylib'> BiocManager::install("PoisonAlien/maftools",force=T)
'getOption("repos")' replaces Bioconductor standard repositories, see '?repositories' for detailsreplacement repositories:CRAN: https://cran.rstudio.com/Bioconductor version 3.16 (BiocManager 1.30.19), R 4.2.2 (2022-10-31)
Installing github package(s) 'PoisonAlien/maftools'
Downloading GitHub repo PoisonAlien/maftools@HEAD
✔ checking for file ‘/private/var/folders/bx/n5y8wbvx6td4rk2q0ht795lm0000gn/T/Rtmp8t6yAm/remotes109d7f7f267f/PoisonAlien-maftools-1c64bf9/DESCRIPTION’ ...
─ preparing ‘maftools’:
✔ checking DESCRIPTION meta-information ...
─ cleaning src
─ checking for LF line-endings in source and make files and shell scripts
─ checking for empty or unneeded directoriesOmitted ‘LazyData’ from DESCRIPTION
─ building ‘maftools_2.12.05.tar.gz’* installing *source* package ‘maftools’ ...
** using staged installation
** libs
clang -mmacosx-version-min=10.13 -I"/Library/Frameworks/R.framework/Resources/include" -DNDEBUG -D_FILE_OFFSET_BITS=64 -I'/Library/Frameworks/R.framework/Versions/4.2/Resources/library/Rhtslib/include' -I'/Library/Frameworks/R.framework/Versions/4.2/Resources/library/zlibbioc/include' -I/usr/local/include -fPIC -Wall -g -O2 -c ntcounts.c -o ntcounts.o
clang -mmacosx-version-min=10.13 -I"/Library/Frameworks/R.framework/Resources/include" -DNDEBUG -D_FILE_OFFSET_BITS=64 -I'/Library/Frameworks/R.framework/Versions/4.2/Resources/library/Rhtslib/include' -I'/Library/Frameworks/R.framework/Versions/4.2/Resources/library/zlibbioc/include' -I/usr/local/include -fPIC -Wall -g -O2 -c snpcounts.c -o snpcounts.o
snpcounts.c:56:26: warning: passing 'const char *' to parameter of type 'char *' discards qualifiers [-Wincompatible-pointer-types-discards-qualifiers]int nloci = countlines(bedfile);^~~~~~~
snpcounts.c:13:22: note: passing argument to parameter 'filename' here
int countlines(char *filename){^
snpcounts.c:56:7: warning: unused variable 'nloci' [-Wunused-variable]int nloci = countlines(bedfile);^
2 warnings generated.
clang -mmacosx-version-min=10.13 -I"/Library/Frameworks/R.framework/Resources/include" -DNDEBUG -D_FILE_OFFSET_BITS=64 -I'/Library/Frameworks/R.framework/Versions/4.2/Resources/library/Rhtslib/include' -I'/Library/Frameworks/R.framework/Versions/4.2/Resources/library/zlibbioc/include' -I/usr/local/include -fPIC -Wall -g -O2 -c somaticfreq.c -o somaticfreq.o
clang -mmacosx-version-min=10.13 -dynamiclib -Wl,-headerpad_max_install_names -undefined dynamic_lookup -single_module -multiply_defined suppress -L/Library/Frameworks/R.framework/Resources/lib -L/usr/local/lib -o maftools.so ntcounts.o snpcounts.o somaticfreq.o /Library/Frameworks/R.framework/Versions/4.2/Resources/library/Rhtslib/usrlib/libhts.a -lcurl -F/Library/Frameworks/R.framework/.. -framework R -Wl,-framework -Wl,CoreFoundation
installing to /Library/Frameworks/R.framework/Versions/4.2/Resources/library/00LOCK-maftools/00new/maftools/libs
** R
** inst
** byte-compile and prepare package for lazy loading
Error in dyn.load(file, DLLpath = DLLpath, ...) : unable to load shared object '/Library/Frameworks/R.framework/Versions/4.2/Resources/library/DNAcopy/libs/DNAcopy.so':dlopen(/Library/Frameworks/R.framework/Versions/4.2/Resources/library/DNAcopy/libs/DNAcopy.so, 0x0006): Symbol not found: (__gfortran_os_error_at)Referenced from: '/Library/Frameworks/R.framework/Versions/4.2/Resources/library/DNAcopy/libs/DNAcopy.so'Expected in: '/Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libgfortran.5.dylib'
Calls: <Anonymous> ... namespaceImport -> loadNamespace -> library.dynam -> dyn.load
Execution halted
ERROR: lazy loading failed for package ‘maftools’
* removing ‘/Library/Frameworks/R.framework/Versions/4.2/Resources/library/maftools’
* restoring previous ‘/Library/Frameworks/R.framework/Versions/4.2/Resources/library/maftools’
Warning message:
In i.p(...) :installation of package ‘/var/folders/bx/n5y8wbvx6td4rk2q0ht795lm0000gn/T//Rtmp8t6yAm/file109d18ef3832/maftools_2.12.05.tar.gz’ had non-zero exit status
这种情况,都删了,然后在官网下载压缩包本地安装一下R包即可
BiocManager::install("DNAcopy", type = "source")