基因组学是生命科学的基础
自从人类基因组计划完成之后,基因组学就成了我们生命科学的基础。就像我们建放房子先修地基一样,地基修好了我们才能不断的去加楼层,在人类基因计划之后又出现了计算基因组学、功能基因组学、比较基因组学、结构基因组学、宏基因组学、营养基因组学等等,这个房子以后会搭的越来越好看。
计算基因组学
计算基因组学是指使用计算和统计分析从基因组序列和相关数据中
破译生物学[10],包括DNA和RNA序列以及其他“后基因组”数据,结合计算和统计方法来理解基因的功能和统计关联分析,该领域通常也被称为计算和统计遗传学/基因组学。
因此,计算基因组学可以被视为生物信息学和计算生物学的一个子集,但重点是使用全基因组(而不是单个基因)来理解物种的DNA如何在分子水平及其他水平上控制其生物学的原理。随着目前大量生物数据集的丰富,计算研究已成为生物发现最重要的手段之一。
功能基因组学
功能基因组学(Functional genomics),功能基因组学是研究基因组的基因和基因间区域如何促成不同的生物过程。功能基因组学侧重于基因产物在特定环境中的动态表达,例如,在特定发育阶段或疾病期间。
比较基因组学:
比较基因组学(Comparative genomics)是基于基因组图谱和测序技术,对已知的基因特征和基因组结构进行比较以了解基因的功能、表达机制和不同物种亲缘关系的生物学研究。
基因组的特征可包括的DNA序列,基因,基因顺序,调控序列,和其它的基因组结构标志。通过对不同亲缘关系物种的基因组序列进行比较,能够鉴定出编码序列、非编码调控序列及给定物种独有的序列。
而基因组范围之内的序列比对,可以了解不同物种在核苷酸组成、共线性关系和基因顺序方面的异同,进而得到基因分析预测与定位、生物系统进化关系等方面的信息。
结构基因组学:
结构基因组学是一门用结构生物学方法研究整个生物体、整个细胞或整个基因组中所有的蛋白质和相关蛋白质复合物的三维结构的学科。
主要利用实验方式(X射线晶体学、核磁共振波谱法和电子显微)来测定蛋白质结构,同时结合同源建模这一计算方式来推测蛋白质结构,和传统结构生物学不同的是,利用结构基因组学所测定的蛋白质结构通常是功能未知的蛋白质,通过三维结构信息来预测蛋白质功能。
结构基因组学重视快速、高通量的蛋白质结构测定。包括三个重要的计划,蛋白质结构启动计划(Protein Structure Initiative, PSI),欧洲结构蛋白质组计划(Structural Proteomics in Europe, SPINE),中国结构基因组计划。
宏基因组学:
微生物在人体的食物消化、机体免疫等方面发挥着重要作用。在大多数情况下,微生物通过群落而非单一个体来发挥这些重要功能。水体、土壤、肠道和很多的人工生物环境(如废水处理、食品发酵、堆肥、沼气池等等)都具有很复杂的微生物群落,这些微生物相互作用、共同协作,一起完成复杂的代谢功能。
环境样品中的微生物组成的群落构成了一个巨大而复杂的基因库,在这个基因库中既包含代表不同微生物身份的系统发育标记基因(如16S rRNA基因),也包含各种代谢功能基因,它们统称为宏基因组(metagenomics,又称宏基因组、环境基因组或生态基因组),这些基因确定了样品微生物群落的组成与功能,研究样品的基因组是认识复杂微生物群落的主要途径。
宏基因组学在开发微生物资源多样性、筛选获得新型活性物质、发掘与抗生素抗性、维生素合成及污染物降解相关的蛋白质等方面展示了很大的潜力。
人类微生物组计划(Human Microbiome Project,HMP)是美国国立卫生研究院(NIH)于2008年发起的一项旨在鉴定与阐明和人类健康与疾病相关的微生物功能的计划。于2007年启动,第一阶段 (HMP1) 专注于识别和表征人类微生物群。第二阶段被称为“综合人类微生物组计划 ”(iHMP),于 2014 年启动,旨在产生资源来表征微生物组并阐明微生物在健康和疾病状态中的作用。
营养基因组学:
20世纪90年代人类基因组计划的启动以及随后的人类DNA测序图谱开创了“大科学时代”,启动了我们今天所知道的营养基因组学(Nutrigenomics)领域[11]。营养基因组学是研究食物如何与基因相互作用,并解释可能影响我们食物中维生素,矿物质和化合物需求的个体遗传差异。每个人都根据其基因组成以不同的方式吸收,代谢和运输化学物质,营养基因组学开启了个体营养图谱的蓝图。
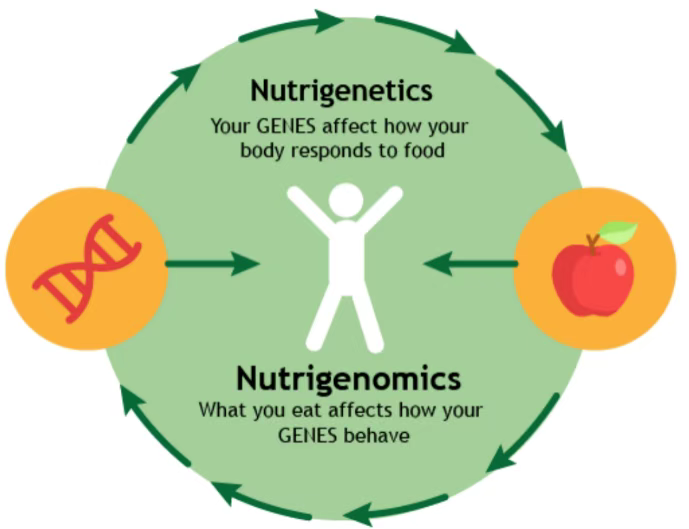
代谢组学:
代谢物是在代谢过程中化学转化的小分子,因此,它们提供细胞状态的功能参数。与功能分别受表观遗传调节和翻译后修饰影响的基因和蛋白质不同,代谢物是生物活性的直接特征,因此它们更容易与表型相关联。代谢组是生物途径的输入和输出的量度,因此,通常被认为比其他组学(如基因组学或蛋白质组学)更能代表细胞的功能状态。
此外,许多代谢物在各种动物物种中都是保守的,有助于将实验动物的研究结果外推到人类。测量代谢组的常用技术包括质谱(MS)和核磁共振波谱(NMR),它能检测数百到数千种独特的化学实体。在这种情况下,代谢组学(metabolomics)已成为一种强大的方法,已被广泛用于临床诊断[12]。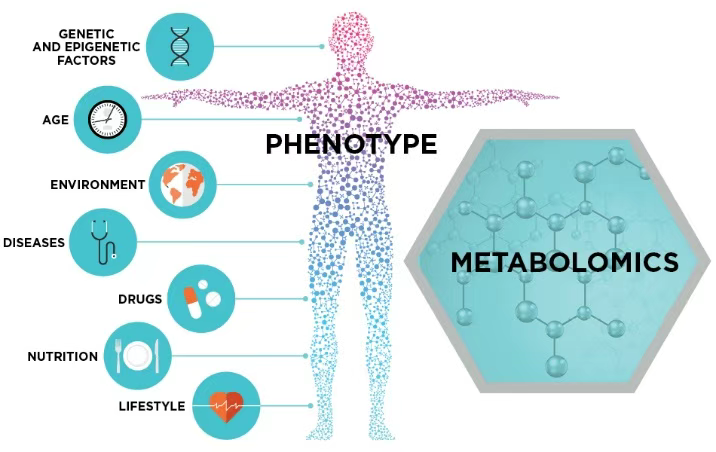
转录组学:
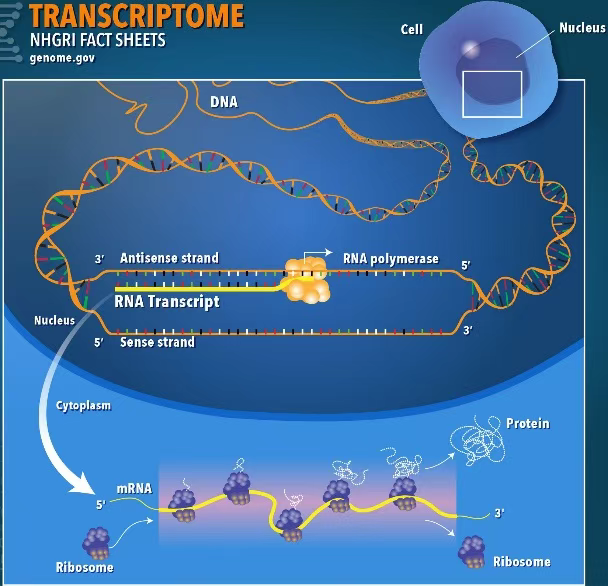
转录组学(Transcriptomics)是对基因型在给定时间产生的RNA转录本的分析,该转录本在基因组,蛋白质组和细胞表型之间提供了联系。转录组是所有RNA分子的集合,包括mRNA,rRNA,tRNA和在一个或一个细胞群中产生的非编码RNA。转录组学也称为表达谱分析,检查给定细胞群中RNA的表达水平[13]。
RNA-Seq一种使用深度测序技术的转录组分析方法,它使用下一代测序(NGS)来揭示特定时间生物样品中RNA的存在和数量,分析不断变化的细胞转录组[14]。
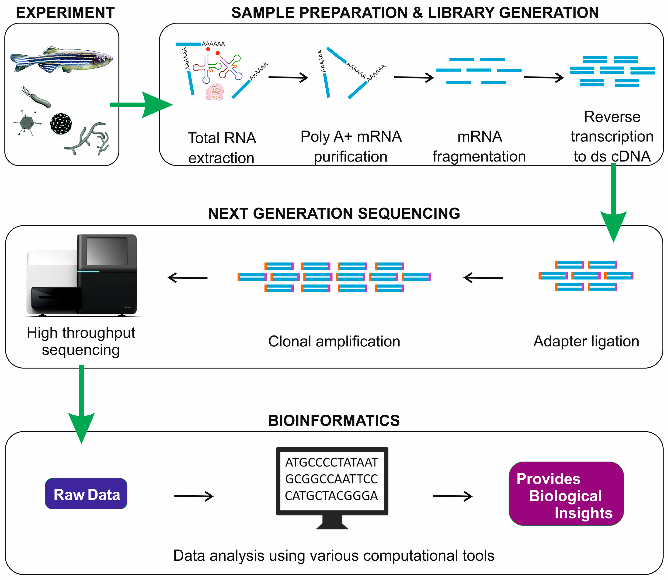
蛋白质组学:
蛋白质组学(Proteomics)是对蛋白质特别是其结构和功能的大规模研究,是在90年代初期,由马克·威尔金斯(Marc Wikins)和学者们首先提出的新名词。它补充了其他“组学”技术,如基因组学和转录组学,以阐明生物体蛋白质特征,并识别特定蛋白质的结构和功能。
它比基因组学更复杂,因为生物体的基因组或多或少是恒定的,而蛋白质组因细胞而异。蛋白质组学研究的关键技术包括质谱分析、X射线晶体学、核磁共振和凝胶电泳。基于不用蛋白质组学的技术用于不同的研究环境,例如检测各种诊断标志物,疫苗生产的候选物,了解致病机制,响应不同信号的表达模式的改变以及解释不同疾病中的功能蛋白途径[15]。
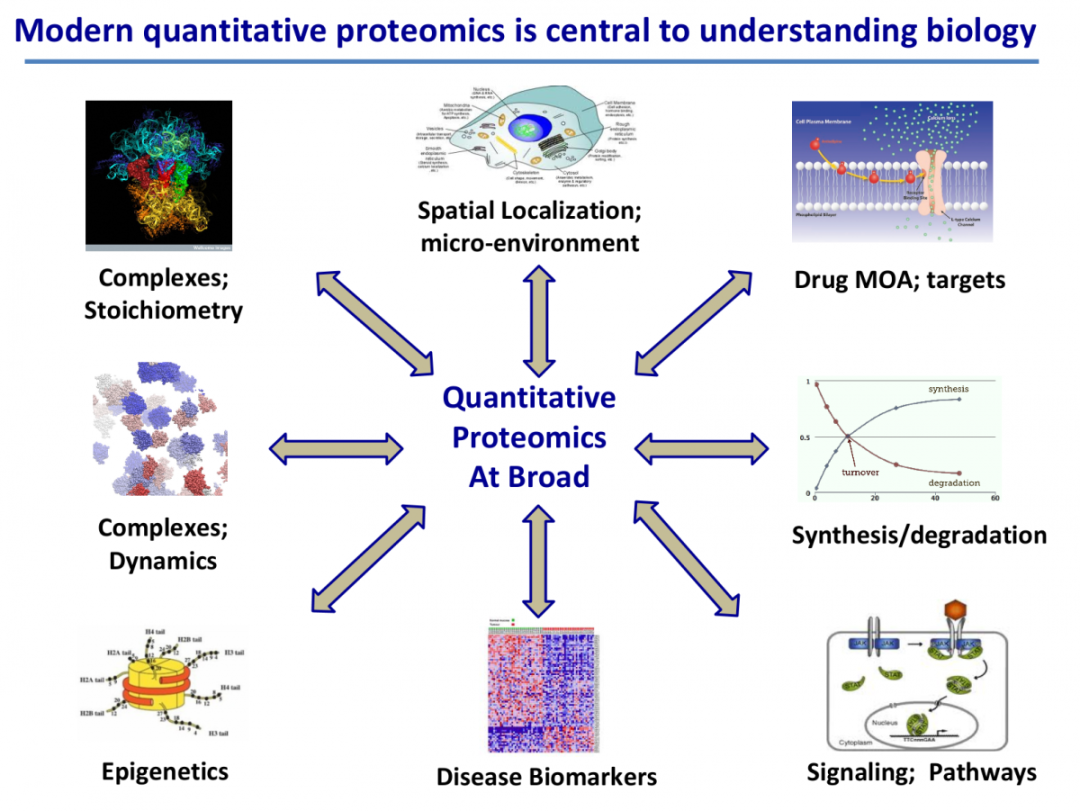
未来发展方向:多组学联合分析
单一的“组学”技术提供了构成细胞,组织和生物体的分子视图。然而,这种观点通常仅限于单个水平,如基因组,转录组,蛋白质组,代谢组等水平。集成单个级别以生成全局视图通常称为多组学方法。多组学方法对于理解癌症等复杂疾病非常有用,其中疾病病因受到多种遗传和环境因素的影响。
多组学方法可以大致分为基于遗传,表型和环境因素的方法[16]。基于基因型的多组学方法旨在使用全基因组关联研究来鉴定与疾病风险相关的位点。进一步检查位点区域可以帮助确定可能在疾病启动中起作用的候选基因。通过探索基因组和转录组水平上的突变或表达变化,可以进一步验证相关基因。
其次,基于表型的多组学方法探索了疾病,临床因素和基于组学的数据之间相关性的知识。第三,基于环境的多组学方法结合了来自组学数据(如微生物组,基因组或代谢组水平)的信息,并估计与吸烟和饮食等环境因素的关联。
多组学提供了一种更全面的方法来解决生物学问题,方法是使用来自不同平台的综合信息在不同维度上观察它。
单细胞转录组:
RNA-seq通常是混合测序,数据代表了数千至数百万个细胞的基因表达模式的平均值;这可能会掩盖细胞之间的生物学相关差异。单细胞RNA-seq(scRNA-seq)代表了一种克服这个问题的方法。通过分离单个细胞,捕获其转录本并生成测序文库,其中转录本被映射到单个细胞,scRNA-seq能够以前所未有的分辨率评估细胞群和生物系统的基本生物学特性[17]。
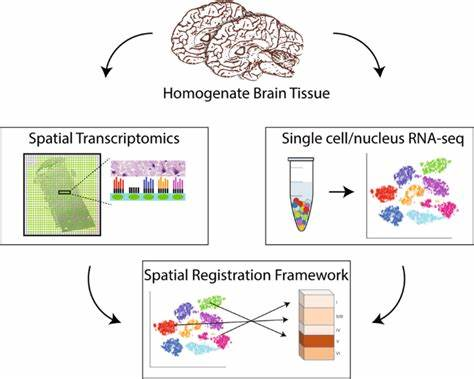
空间转录组:
空间转录组学是一系列方法的总体术语,旨在将细胞类型(由mRNA读数识别)分配到它们在组织学切片中的位置[18]。
空间转录组测序的工作流程主要分为两部分:组织学部分和组学部分。组织学部分包括样品包埋、切片、固定、染色和成像,并记录切片的形态学信息;组学部分包括cDNA合成、扩增、接合和测序,并记录该部分的转录本信息和空间位置信息。
使用10X Genomics空间转录组测序技术,用于文库构建的每张载玻片都有四个捕获区域,其中每个捕获区域包含5000个条形码斑点,每个斑点具有唯一的条形码序列。组织部分的细胞会释放mRNA,迁移到每个斑点的mRNA会用相应的条形码序列标记,然后构建和测序文库。
最后,根据数据的条形码信息对数据进行分析,确定哪些数据来自哪个位置,从而实现空间基因表达的可视化。