DrugGPT chatgpt druggen 之间的关系 ,DrugGPT是基于gpt2基础上开发的
DrugGPT chatgpt druggen 之间的关系 ,DrugGPT是基于gpt2基础上开发的
# GPT-4 是否开源的现状分析
结合现有信息,OpenAI 官方的 GPT-4 核心版本仍为闭源状态,但存在两类与“GPT-4 开源”相关的关键情况:一是社区开发的开源替代方案,二是 OpenAI 推出的开源模型(非 GPT-4 本体),具体如下:
一、官方 GPT-4:闭源属性未改变,核心信息不公开
OpenAI 自 2023 年发布 GPT-4 以来,始终未开放其核心模型的代码、训练数据、模型权重或架构细节,且这一状态在后续多年未发生本质变化:
- 技术细节严格保密:根据 2023 年 3 月的报道(摘要 5),OpenAI 仅发布过一份 98 页的“技术报告”,但明确拒绝披露 GPT-4 的架构(如模型参数规模)、训练硬件、计算资源消耗及数据集构建方式,理由涉及“竞争格局”与“安全影响”。这种保密程度被研究者批评为“违背科学透明性”,甚至被评价为“产品而非科学”。
- 无官方开源计划或行动:截至 2025 年 10 月的最新信息(如摘要 2、4、6),OpenAI 从未宣布“开源 GPT-4”,其闭源属性导致开发者无法自主复现 GPT-4 性能、创建衍生模型或独立部署,只能通过官方 API 调用(且需支付费用)。
二、“开源 GPT-4”的两种常见误解:替代方案与新开源模型
当前市场上被提及的“开源 GPT-4”,实际多指向两类内容,而非官方 GPT-4 本体:
(一)社区开发的“GPT-4 开源替代模型”:功能相似但非同源
为弥补 GPT-4 闭源的局限,开源社区推出了多款性能接近 GPT-4 的替代模型,这些模型具备开源属性(可获取代码、权重),但与 OpenAI 的 GPT-4 无技术继承关系:
- 典型代表与特性:
- ColossalChat/FastChat(摘要 1、3):基于完整的“人类反馈强化学习(RLHF)”流程开发,提供训练代码、双语数据集及 4 位量化推理方案,支持低成本定制聊天机器人,其衍生模型 Vicuna 被宣称能达到 ChatGPT(GPT-3.5 级)90% 的性能,可作为 GPT-4 的轻量化开源替代。
- GPT4All(摘要 1):基于 LLaMA 架构,训练数据包含大量对话、代码、故事等交互内容,支持 CPU/GPU 低延迟推理,提供 Python 客户端、LangChain 后端及聊天界面,适合本地部署,定位为“平民化 GPT-4 替代工具”。
- 开源代码解释器(摘要 4):可模拟 GPT-4 代码解释器的核心功能(如安装 Python 库、本地终端运行、联网能力),若无 OpenAI API,还可替换为 Code LLaMA 等开源模型,实现“类 GPT-4 代码生成与执行”。
- 共性特点:这类模型均通过自主训练实现“接近 GPT-4 的功能”,开源且资源需求更低(部分支持 CPU 运行),但性能通常略逊于官方 GPT-4,且部分模型受非商业许可限制(仅允许研究/学术使用)。
(二)OpenAI 2025 年推出的“GPT-OSS”:开源但非 GPT-4
2025 年 8 月,OpenAI 发布了首款开源权重模型系列 GPT-OSS(摘要 6),易被误认作“开源 GPT-4”,但两者存在明确区别:
- GPT-OSS 的核心属性:
- 包含 gpt-oss-120b、gpt-oss-20b 两个版本,采用 Apache 2.0 协议(允许免费商用、微调、嵌入自研产品),原生支持 128K 长上下文、函数调用、推理链输出等高级功能。
- 性能对标 OpenAI 闭源的“GPT-4 轻量化版本”(如 o4-mini、o3-mini),而非完整版 GPT-4,定位是“低门槛开源模型”,旨在推动 AI 普及,而非替代 GPT-4。
- 与 GPT-4 的关键差异:GPT-OSS 是 OpenAI 全新推出的开源产品线,架构为“纯文本推理型”,未继承 GPT-4 的完整能力(如多模态理解、复杂逻辑推理),本质是“OpenAI 开源模型”而非“开源的 GPT-4”。
三、总结:明确“开源”边界,按需选择工具
- 核心结论:官方 GPT-4 未开源,其代码、权重、训练细节仍完全保密;市场上的“开源 GPT-4”要么是社区开发的替代模型(功能相似、非同源),要么是 OpenAI 新推出的 GPT-OSS(开源但非 GPT-4)。
- 选择建议:
- 若需“完全匹配 GPT-4 性能”:只能通过 OpenAI 官方 API 调用,无开源选项。
- 若需“低成本开源方案”:可选择 ColossalChat、GPT4All 等社区模型,适合研究、本地部署或非商业场景。
- 若需“商用友好的 OpenAI 开源模型”:可优先考虑 GPT-OSS(Apache 2.0 协议),性能接近 GPT-4 轻量化版本,且无商用许可限制。
DrugGPT所用GPT版本分析
结合现有文档及补充搜索信息,DrugGPT的核心GPT版本关联可从基础架构参考与专项优化版本两个维度明确,具体如下:
一、核心基础架构:基于GPT-2架构开发
根据最初文档中对DrugGPT的技术描述,其核心架构明确基于GPT(Generative Pre-trained Transformer)系列的基础版本,且进一步指向GPT-2架构。文档提到“DrugGPT, an LLM based on the generative pre-trained transformer (GPT) architecture”,同时在DrugGen(基于DrugGPT优化)的开发过程中,采用了与GPT-2兼容的字节对编码(BPE)分词器(53,083 tokens),并遵循GPT系列“自回归生成序列”的核心逻辑——这与GPT-2的技术特征高度匹配,且未提及采用GPT-3或更高版本的架构特性(如更大参数规模、多模态支持等),因此可确定DrugGPT的基础架构源自GPT-2,通过针对药物分子生成场景(如SMILES字符串、蛋白序列匹配)进行定制化训练,而非直接采用GPT-3/4等更高阶的通用GPT版本。
二、专项优化版本:存在“Drug-GPT™4”等场景化衍生版本
补充搜索的arXiv论文(摘要1)提及“Drug-GPT™3”和“Drug-GPT™4”两个专项版本,这两个版本是在基础GPT架构(推测仍以GPT-2为底层)上,针对医疗健康场景(如特应性皮炎患者体验、糖尿病医护讨论) 进行二次优化的衍生版本:
- 核心差异在于训练数据与场景适配:Drug-GPT™3/4通过整合“患者和医护人员(HCP)的社交媒体、留言板帖子”等医疗专项数据集,强化了在特定疾病领域的问答能力,例如生成更精准的特应性皮炎患者体验相关信息、糖尿病诊疗讨论内容;
- 与通用GPT版本的区别:这类“Drug-GPT™X”版本并非OpenAI官方发布的GPT-3/4,而是开发者基于基础GPT架构(如GPT-2),针对医疗细分场景优化后的“场景化衍生版本”——其命名中的“3/4”更偏向于开发者内部的版本迭代编号,而非对应OpenAI GPT-3/4的技术层级,本质是通过专项数据微调实现“领域适配”,而非采用更高阶GPT的核心架构。
三、总结:DrugGPT的GPT版本定位
DrugGPT的GPT版本需区分“基础架构”与“场景化迭代”:
- 基础技术架构:基于GPT-2开发,核心逻辑为“自回归生成序列”,通过适配药物分子数据(SMILES、蛋白序列)完成定制化训练,是“通用GPT-2架构+药物场景微调”的结合体;
- 场景化衍生版本:存在“Drug-GPT™3/4”等编号版本,属于在基础架构上针对医疗细分场景(如特定疾病问答)进一步优化的迭代版本,命名中的“3/4”是场景化优化的版本号,与OpenAI官方GPT-3/4无直接技术关联。
简言之,DrugGPT未采用GPT-3及以上的通用GPT版本,而是以GPT-2为基础架构,通过“基础架构固定+场景数据微调”的模式,形成适用于药物生成(DrugGPT)或医疗问答(Drug-GPT™3/4)的专项模型。
由牛津大学AI医疗实验室开发的DrugGPT,正是人工智能技术在减少用药错误领域的一次创新尝试。这款AI工具旨在为医生和患者提供用药决策的“安全网”,通过快速生成“第二意见”,帮助医疗专业人士更准确地开具处方。
通过输入患者的病情,DrugGPT可以即时提供推荐药物,同时标注可能的副作用和药物间的相互作用。与其他AI工具不同,DrugGPT的突出特点在于其透明性:不仅提供建议,还详细说明推荐背后的逻辑和参考依据,包括相关研究和临床指导。这种透明度让医生能够审慎地评估和比较建议,在人工智能的辅助下做出更科学的用药决策。
牛津大学AI医疗实验室的负责人大卫·克利夫顿教授(David Clifton)表示:“我们开发的这款工具,不仅是一个助手,更像是医生的‘副驾’。它可以帮助医生与自身的决策进行对比,但决策权始终掌握在人类手中。”
专业表现可媲美医生,但人类仍是关键
根据DrugGPT团队发布的研究预印本,这款AI工具在美国医学执照考试中的表现与人类专家不相上下,这显示了它在复杂医疗场景中的可靠性和精准度。然而,克利夫顿教授强调,DrugGPT的目标并非取代医生,而是通过提供建议和信息支持临床判断。“电脑说不”的机械化决策并不适合医疗领域。
皇家全科医师学院(Royal College of GPs)副主席迈克尔·穆尔霍兰博士(Michael Mulholland)同样表示:“尽管绝大多数情况下医生的处方是正确的,但在高强度工作负荷下,尤其是面对需要同时服用多种药物的患者时,错误可能难以完全避免。引入更先进的安全措施有助于进一步减少人为错误,但我们必须确保新工具在全面推广前经过严格测试和试点,以避免意外后果。”
提升患者用药依从性与理解力
用药错误并不仅仅局限于医生环节。患者因未按医嘱服药导致的“用药不依从”现象,每年给英国国家医疗服务体系(NHS)带来高达3亿英镑的浪费。DrugGPT在这方面也有潜力发挥作用。
牛津大学医院NHS基金会信托顾问医生露西·麦基洛普(Lucy Mackillop)博士指出,DrugGPT通过提供更多药物信息,可以帮助医生与患者进行更有效的沟通,增强患者对药物的理解。“当患者明白用药的必要性和效果时,他们更可能遵从医嘱,这也更有利于药物的效果发挥。”
技术落地需谨慎推进
尽管DrugGPT的潜力令人期待,但AI工具的实施并非一帆风顺。穆尔霍兰博士提醒道,除技术本身外,支持AI工具顺利运转的基础条件也至关重要,例如为全科医疗提供充足的资金和人力支持。此外,在部署过程中,必须确保系统的稳健性,并通过试点发现并解决潜在问题。
人工智能与医疗的未来
DrugGPT的推出标志着人工智能在减少用药错误、提高患者安全方面迈出了重要一步。作为医生的“副驾”,它提供了智能化、数据驱动的支持,同时保留了人类决策的核心地位。
随着技术的不断发展,AI与医疗专业知识的结合或将为医疗行业带来更多突破。从DrugGPT的成功经验来看,未来更多此类工具可能进一步提高医疗服务的质量和效率,为患者带来切实的健康收益。
人工智能的力量不在于取代人类,而在于协同创新。在医疗领域,这种“人机共生”的合作模式无疑是变革的关键。
drugGPT快速探索化合物空间助力药物研发
今天我们介绍由西安交通大学第一附属医院的Yuesen Li发表在BioRxiv上的工作。该工作提出了一种基于自回归模型GPT的配体设计策略DrugGPT,该模型专注于化学空间的探索和特定蛋白质的配体发现。深度学习语言模型在蛋白质设计和生物医学文本分析等领域展示了显著的潜力,为DrugGPT的提出提供了有力支持。在本文中,研究者采用DrugGPT模型学习了大量的蛋白质-配体结合数据,旨在发现能够与特定蛋白质结合的新型分子。这一策略不仅极大地提高了配体设计的效率,还为药物开发过程提供了快速有效的途径,为制药领域带来了新的可能性。
背景介绍
在过去几十年中,计算化学和生物信息学取得了显著的进展,对药物发现领域产生了重大影响。然而,揭示和开发新型药物的旅程仍然面临巨大的挑战和高昂的成本。其中一个主要挑战是化学空间的庞大程度。从理论上讲,潜在的药物样化合物的数量接近无限,使得对这个化学空间的全面和有效探索变得异常困难。
尽管传统的计算机辅助药物设计策略,如分子对接、定量构效关系(QSAR)、药效团建模、无监督学习、深度学习和生成对抗网络(GANs)在一定程度上缓解了这个问题,但对这个巨大化学空间更有效的探索需求需要发展创新的方法和策略。
近年来深度学习技术的出现在药物发现领域引入了新的机会。例如,Atomwise公司利用基于深度卷积神经网络的AtomNet技术进行药物发现。Insilico Medicine公司成功地利用GANs和深度学习方法生成潜在的药物活性候选化合物。与此同时,学术界也报道了基于深度学习的药物发现模型取得了成功,例如由Gupta等人研究的生成递归网络,由Méndez-Lucio等人介绍的基于人工智能的药物活性分子生成方法,以及由Popova等人开发的深度强化学习方法。尽管取得了这些成功,化学空间的庞大仍然需要更有效的解决方案。
为了更有效探索化学空间,本文提出了DrugGPT模型,在DrugGPT模型中对蛋白质和配体进行了分词。例如,在考虑了包含超过20亿化合物的ZINC20数据库之后,本文发现只需要5,373个标记就能准确表示这些化合物,这是通过应用字节对编码(BPE)算法得出的。这一发现表明,尽管可能的化合物数量接近无限,但表示这些化合物的词汇是有限的。通过使用DrugGPT进行学习,可以掌握这些有限词汇的组合和排列方式,从而有效地探索这个广阔的化学空间。
DrugGPT模型采用自回归生成的方法,这种方法利用模型的输出作为输入进行预测,增强了捕捉化学结构和活性关系的准确性,并提高了生成化合物的质量。通过自回归生成方法,模型表现出更强的稳定性,在训练过程中更容易进行优化和调整。
图1:使用DrugGPT的示意图。第一步是“选择输入类型”,用户有“蛋白质氨基酸序列”、“蛋白质氨基酸序列+配体提示”和“无”三种选择。一旦选择了适当的输入类型并提供了相应的输入,就会生成输入原始文本。第二步涉及使用DrugGPT标记器来处理输入的原始文本,从而形成input id。在第三步中,将输入id输入到DrugGPT模型中,生成输出。在最后一步,这个生成的输出进行后处理,最终产生预测的药物。
本文的创新点与贡献:
通过重新设计分词器和从头重新训练,优化了GPT-2模型,生成了专注药物设计任务的DrugGPT模型。
DrugGPT能更准确地捕捉药物分子与蛋白质结合信息,有效生成潜在活性药物。
DrugGPT采用最大化条件概率和反向传播训练,并具有强大的泛化能力。
实验结果
配体的标记化
本文采用了字节对编码(BPE)算法对来自两个数据库的配体进行标记化。考虑到ZINC20数据库中的20亿个化合物,本文发现只需要5373个标记就能表示这些分子。这意味着BPE算法可以用有限的词汇高效表示大量化合物。本文还分析了已经与蛋白质结合的配体,这些配体来自另一个数据库。在应用BPE算法标记这些配体后,发现3560个标记足以准确描述这些已经与蛋白质结合的配体。这3560个标记不仅可以表示第二个数据库中的配体,还可以覆盖ZINC20数据库分子,以及描述可开发成药物的分子。在DrugGPT训练的第一阶段,本文使用这3560个标记来表示化合物,不仅增强了模型表示实际配体能力,还降低了计算复杂性。实验结果表明,使用从已知蛋白质相互作用的配体中提取的缩减词汇表,可以增强药物发现效率和适用性。
蛋白质的标记化
本文也应用BPE算法对蛋白质进行标记化。但与配体不同,蛋白质的复杂性远超过配体。在对一个蛋白质数据库中的1.8万多个蛋白质序列使用BPE算法后,生成了130多万个蛋白质标记。这说明蛋白质氨基酸序列比配体更加复杂。考虑到自然界中的2000万个蛋白质,如果都应用BPE算法,生成的词汇表将非常庞大。从生成的词汇表大小的角度来看,大约每两个蛋白质序列产生一个标记,进一步证明了蛋白质序列的复杂性。本文借鉴了GPT-2模型,设置蛋白质的词汇表大小为5万。同时本文使用“
+蛋白质序列++配体SMILES”表示文本,然后进行BPE标记化。由于蛋白质比配体更复杂,需要更大的词汇表来表示。这种方法还避免了重复蛋白质的出现。总之,本文使用BPE算法对蛋白质进行了标记化,并设置了相对较大的词汇表大小,以更好地表示蛋白质序列的复杂性。这一步为后续DrugGPT模型的训练提供了重要的数据基础。
构建DrugGPT标记器
在构建DrugGPT的词汇表时,本文首先对配体和蛋白质的词汇表进行了并集操作。由于SMILES表示和氨基酸序列中存在重叠的字符,这意味着这两个词汇表中存在相同的标记。在合并这些词汇表时需要处理这些重复的标记。BPE标记器包括两个文件:vocab.json(用于存储标记)和merges.txt(用于记录标记的合并操作)。在配体和蛋白质的标记器中检查了这两个文件,并且删除了相同的标记和具有不同合并操作的相同标记。
图2:DrugGPT标记器的构建过程。DrugGPT标记器是基于Hugging Face的jglaser/binding_affinity数据库构建的。首先,使用BPE算法对数据库中的蛋白质氨基酸序列和配体SMILE进行处理,创建蛋白质标记器。随后,使用BPE算法对数据库中的配体SMILES进行类似处理,形成配体标记器。最后,合并蛋白质标记器和配体标记器,删除两者中存在的任何重复标记,从BPE算法中排除初始标记。
在BPE标记化算法后删除了重复的标记,然后使用初始的256个字符来补充标记,弥补重复标记删除造成的缺口。通过这些步骤,成功构建了包含53080个标记的DrugGPT标记器。在标记化前,原始文本的平均长度为700,90%不超过1213。但在标记化后,序列长度减小,平均长度降至200,只有1.3%超过768。这表明标记后的长度与原始文本长度有更合理的分布,有助于DrugGPT模型的训练。总的来说,通过合并蛋白质和配体的词汇表、处理重复标记和补充字符,成功构建了DrugGPT的标记器。这不仅有效地整合了词汇表,还导致序列长度有更合理的分布,为后续模型的训练和应用奠定了重要的基础。
图3:DrugGPT标记器减少了序列的长度。(a)和(b)分别描绘了获得蛋白质配体原始文本之前配体SMILES长度和氨基酸序列长度的频率直方图。(c)描述了原始文本的文本长度与频率的关系,原始文本是对jglaser/binding_affinity数据库中的数据进行’’ +’
’ +蛋白质序列+‘’ +配体的SMILES序列+''处理后得到的。(d)说明了输入id的令牌长度和频率之间的关系,这些id是在使用DrugGPT令牌器处理(c)中的原始文本后获得的。通过比较(c)和(d)可以观察到,在使用DrugGPT标记器处理后,标记的长度明显小于原始文本的长度。这大大减少了输入到DrugGPT模型的长度,从而大大减少了训练的计算开销。
训练DrugGPT模型
图4:DrugGPT的构建过程。每个epoch内的DrugGPT训练分为两个步骤。第一步,数据来源于ZINC20,用ZINC20生成相应的配体原始文本。然后使用DrugGPT标记器派生配体输入id,使模型能够学习如何准确地表示化合物并了解其固有的化学结构和性质。第二步使用来自jglaser/binding_affinity数据库的数据来创建相应的蛋白质配体原始文本。再次使用DrugGPT标记器,获得蛋白质配体输入id,从而教会模型如何为特定蛋白质生成相应的化合物。一旦设置了训练参数,GPT2模型就会从头开始训练,以生成DrugGPT模型。
在训练DrugGPT模型时,本文使用了Hugging Face提供的强大NLP库transformers和datasets,这极大提高了效率。选择了GPT2LMHeadModel作为基础模型,并且采用从头开始训练,而非微调预训练模型,这是为了更好捕捉配体和蛋白质的结构信息。总共设计了两个阶段训练:第一阶段以配体文本训练,使模型学习表示配体和理解化学结构。第二阶段以蛋白质-配体对文本训练,使模型能为蛋白质生成相应配体。经过五个训练时期,模型损失在验证集上降至0.04,这表明模型掌握了有用信息。使用NVIDIA RTX 4090显卡来加速训练,同时设置了AdamW优化器和合理的超参数,最后成功训练出了DrugGPT模型。结果表明,模型已经准备好在配体发现领域提供帮助。这为药物发现领域的进一步研究与实践奠定了坚实的基础。简单来说,通过使用NLP库、从头训练、分阶段训练、硬件加速、合理超参数等方法,成功训练出了DrugGPT模型,这为后续药物发现工作提供了支持。
模型推理和配体设计策略
DrugGPT模型被用于探索配体设计的潜能。设计了三种推理方法用于设计蛋白质的配体:
只基于给定蛋白质序列
基于给定蛋白质序列并提供配体SMILES格式信息作为提示
不提供任何信息,允许模型自主生成配体在第一种方法中,直接输入蛋白质氨基酸序列或FASTA格式;在第二种方法中,提供配体SMILES格式的信息作为提示;在第三种方法中,不提供任何信息,观察模型自主生成的配体方案。DrugGPT Notebook 和命令行界面实现了以上三种推理模式。三种推理方法可根据需求选择,完成不同蛋白质的配体设计。简单来说, 通过提供不同类型的信息作为输入,采用不同的推理方法,设计适用于各种蛋白质的配体。
BCL-2蛋白质的配体设计
本文选择BCL-2作为一个重要的抗癌药物靶点,以展示配体设计模型DrugGPT在探索潜在抗癌配体方面的能力。BCL-2是一种抗凋亡蛋白质,通过抑制细胞凋亡过程促进肿瘤细胞的存活。BCL-2在许多类型的癌症中发挥关键作用,特别是在慢性淋巴细胞性白血病(CLL)和非霍奇金淋巴瘤等血液肿瘤中。因此,BCL-2被认为是一个具有重要治疗价值的潜在抗癌药物靶点。本文使用以下命令开始模型的推理过程:python drug_generator.py -f bcl2.fasta -n 50。该命令指示模型为BCL-2生成至少50个可能的配体,并以sdf格式返回它们的三维结构。最终,模型成功返回了73个可能的配体。这些配体的SMILES表示通过以下20个示例进行展示,如下所示:
表1:DrugGPT生成的对于BCL-2的20个潜在配体的SMILE表示
本节的实验结果展示了DrugGPT在为特定蛋白质设计配体方面的能力,为药物发现领域的未来研究提供了新的工具和方法。
具有针对BCL-2蛋白质的配体提示的配体设计
本文引入了一个新颖的概念,称为“配体提示”,以实现对配体结构的定制化。本质上,这个提示是用户希望包含在配体的SMILES表示中的特定初始部分。使用这种策略,本文可以为特定的蛋白质设计配体,并指定它们的初始部分,使得对特定化学基团的调整和优化成为可能。为了验证这种策略,本文设定了一个目标,即设计以“COc1ccc(cc1)C(=O)”开头的配体。本文执行了以下命令:python drug_generator.py -f bcl2.fasta -l COc1ccc(cc1)C(=O) -n 50,以生成至少50个具有这个特定结构作为起始部分的配体。基于BCL-2蛋白质的FASTA序列和指定的SMILES起始部分,这个命令成功生成了54个潜在的配体。下表展示了20个选定配体的SMILES表示:
表2:基于BCL-2 Fasta序列和指定起始部分的DrugGPT生成的20种潜在配体的SMILE表示
所有生成的配体都以“COc1ccc(cc1)C(=O)”开头,这证明该配体提示策略可以成功定制特定的化学基团。用户可以根据需要进一步调整这个起始化学基团后面的分子结构来优化配体,或直接创建一个新的配体。上述过程演示了如何使用配体的SMILES表示和用户指定的起始结构(即配体提示)为特定蛋白质生成配体。这种方法既灵活又可定制,为配体设计提供了一种有效的策略。
直接推理的应用:ENPP2配体设计
本文采用了一种有趣的方法,称为直接推理,该方法反映了模型在学习近190万个蛋白质-配体对之后最有可能输出的结果。这种推理显示了DrugGPT模型倾向为哪些蛋白质生成哪些配体。在本次运行中,设置参数为至少返回200个配体,最终返回了201个配体,但在生成这201个配体时保留了1061个直接推理方案。分析这些方案,本文发现最多的设计配体是为具有以下序列的蛋白质设计的:
对该序列进行BLAST分析得到以下结果:
图5:BLAST结果为直接推断结果中为其设计的配体最多的蛋白质。BLAST分析表明该序列与ENPP2的氨基酸序列一致。
这表明DrugGPT模型极度倾向于为ENPP2设计配体。在最终返回的201个配体中,有167个配体是为ENPP2设计的。随后,本文登录BindingDB并下载了当前数据库中与ENPP2相关的所有2685个配体。在这些设计的配体中,有112个与现有配体匹配,55个是完全新设计的配体,不包含在这2685个配体中。这112个匹配配体可以理解为DrugGPT模型学习到了这些蛋白质-配体文本,并且能够再现这些蛋白质-配体文本,这证明本文对DrugGPT模型的训练是充分的。这55个新设计的配体分子表明,即使在学习了大量ENPP2配体分子之后,模型仍然可以避开众多现有的配体分子,创造性地设计全新的配体分子,展示了训练有素的DrugGPT模型的创造力。此外,本文还注意到ENPP2在癌症、炎症性疾病和纤维化等多种疾病中发挥着重要的生物学作用,发现更有效的ENPP2抑制剂具有重要的临床意义。然而,当前针对ENPP2的药物选择仍然有限,许多药物在疗效、安全性和副作用方面需要进一步优化。在这种情况下,本文的研究揭示了DrugGPT模型在药物开发领域的巨大潜力。
生成配体后的后处理
图6:生成配体的后处理程序。最初,使用DrugGPT标记器对模型输出进行解码,从而得到预测的文本。随后,对预测文本进行拆分,提取“”之后的部分,表示配体的SMILES。最后,Open Babel用于筛选和转换配体的SMILES格式。
在生成配体后,本文对SMILES字符串进行筛选排除不合理配体,将通过条件筛选的SMILES转换为SDF格式并生成3D构象。这可以确保文件名不受限制,同时也可以通过哈希值追溯原始SMILES。使用分子可视化软件查看生成的SDF文件并且从立体化学、合成可行性、相似性和药物样性几个方面评估生成的配体结构。即使生成的配体结构不合理,也可以分析并应用于现有药物。对于合理的配体,本文使用分子对接、QSAR分析和药效团分析方法筛选和优化。这些方法可以评估配体的活性和药物样性。在筛选和优化后,本文对选择的化合物进行实验验证以评估它们对目标蛋白的实际活性。这提供了完整的配体筛选和优化流程有助于发现新型配体。
讨论
通过配体标记化有效地探索化学空间
通过将配体的化学结构转换为标记序列,这一过程类似于将长句子分割成单词。这种标记化策略不仅简化了化合物的表示,而且使能够以与处理文本相同的方式处理化学结构。与逐字解释SMILES表示相比,使用训练过的标记来表示SMILES更有效。在处理来自ZINC20库的超过20亿个分子后,BPE算法收敛,产生仅包含5373个标记的词汇表。在探索化学空间的过程中,可以利用配体的标记序列生成新的化合物,这一过程类似于语言模型在给定上下文下生成新句子的过程。通过控制生成过程中的随机性,既可以保证生成化合物的质量,又可以实现对化学空间的广泛探索。此外,还可以根据蛋白质结构信息调整化合物生成策略。该方法不仅提高了药物发现的效率,而且为药物设计提供了一种新的途径。在未来的研究中,将进一步优化模型,以更好地服务于药物发现任务。
在标记化的镜头下,从蛋白质和配体之间的复杂性差异洞察药物发现
蛋白质的词元数量大于配体词元数量,这表明蛋白质中氨基酸的排列具有更高的复杂性。这种复杂性给蛋白质靶点鉴定和药物设计带来了重大挑战。此外,蛋白质中的平均氨基酸序列长度大于配体中SMILES表示的长度。这可以通过一个语言上的类比来更好地理解这个问题——想象一个词汇量有限的语言试图用词汇量更大的语言来表达概念。在有限的语言中找到合适的单词来表达丰富含义可能具有挑战性。同样,在表示蛋白质和配体时,蛋白质具有更大的词汇量,这意味着需要更多的符号来表示蛋白质的结构和功能。在实际的药物设计中,必须在有限的配体词汇中找到能够有效描述与蛋白质相互作用的化合物。虽然BPE算法确实可以表示蛋白质,但蛋白质序列的高复杂性会导致在标记化过程中出现大量的标记数量。同时,有限数量的配体标记可以有效表示大量的化合物。在未来的研究中,需要找到更有效的解决这些问题的方法,以提高药物开发的准确性和效率。可能需要改进BPE算法以更好地捕获蛋白质和配体之间的特征,或开发新的表示方法以更有效地描述蛋白质-配体相互作用。
自回归生成与对抗生成的优势比较
在图像生成领域,生成对抗网络GAN相较自回归模型GPT可能生成效果不稳定、训练过程不稳定。相比之下,GPT在图像生成任务中表现更好。基于GPT-3的图像生成模型DALL-E能通过文本输入生成对应图像。自回归模型可更有效处理文本生成任务中的挑战。而GAN在此存在一定难度。自回归模型(如GPT)使用SMILES符号生成药物分子,能更高效捕获分子信息。推测基于自回归模型的药物设计相较GAN有更高生成质量、更稳定训练过程和更强泛化能力等优势。基于自回归模型的药物设计方法能帮助研究人员更快发现活性候选分子,提高药物研发效率和准确性。期望自回归模型在未来药物研发将发挥更大作用,为人类健康福祉做贡献。
参考资料
Li, Yuesen, Chengyi Gao, Xin Song, Xiangyu Wang, Yungang Xu, and Suxia Han. “DrugGPT: A GPT-based Strategy for Designing Potential Ligands Targeting Specific Proteins.” bioRxiv (2023): 2023-06.
本文档聚焦于新型大语言模型 DrugGen 的研发、优化及性能评估,旨在解决传统药物设计及现有模型(如 DrugGPT)的局限性,推动药物研发效率提升。
传统药物设计因化学和生物复杂性高,临床试验失败率高,耗时且成本高昂;虽深度生成模型(如 DrugGPT)为药物设计提供新思路,但 DrugGPT 存在生成化学结构有效性不足(仅 95.5%)、未整合已批准药物特征、生成分子结合亲和力较低(5.81 [4.97–6.63])等问题。
为此,研究团队以 DrugGPT 为基础开发 DrugGen:通过在已批准药物 - 靶点相互作用数据集上进行监督微调(SFT),结合近端策略优化(PPO)强化学习算法,并引入基于预训练转换器的蛋白质 - 配体结合亲和力预测模型(PLAPT)和定制化无效结构评估器提供奖励反馈,实现模型优化。
性能评估显示,DrugGen 在多维度表现优异:化学结构有效性达 100%,生成分子结合亲和力显著提升(7.22 [6.30–8.07]),同时保持结构多样性与新颖性;分子对接模拟验证其有效性,如针对脂肪酸结合蛋白 5(FABP5)生成分子的对接分数(-9.537、-8.399)优于参考分子棕榈酸(-6.177),针对 NAMPT 生成分子对接分数(-8.381)优于参考分子 Daporinad(-8.3)。此外,DrugGen 还具备药物重定位潜力,能为现有靶点创建新型药效团,不过存在靶点间结合亲和力结果差异(如 FABP5 性能提升不显著)、无法精准靶向特定结合位点(如 ACE 多结合位点问题)、依赖的亲和力预测模型有精度局限、未生成完全优化结构等局限性,未来需通过优化奖励函数、整合其他模型(如 PocketGen)、结合实验验证等方式改进。
二、脑图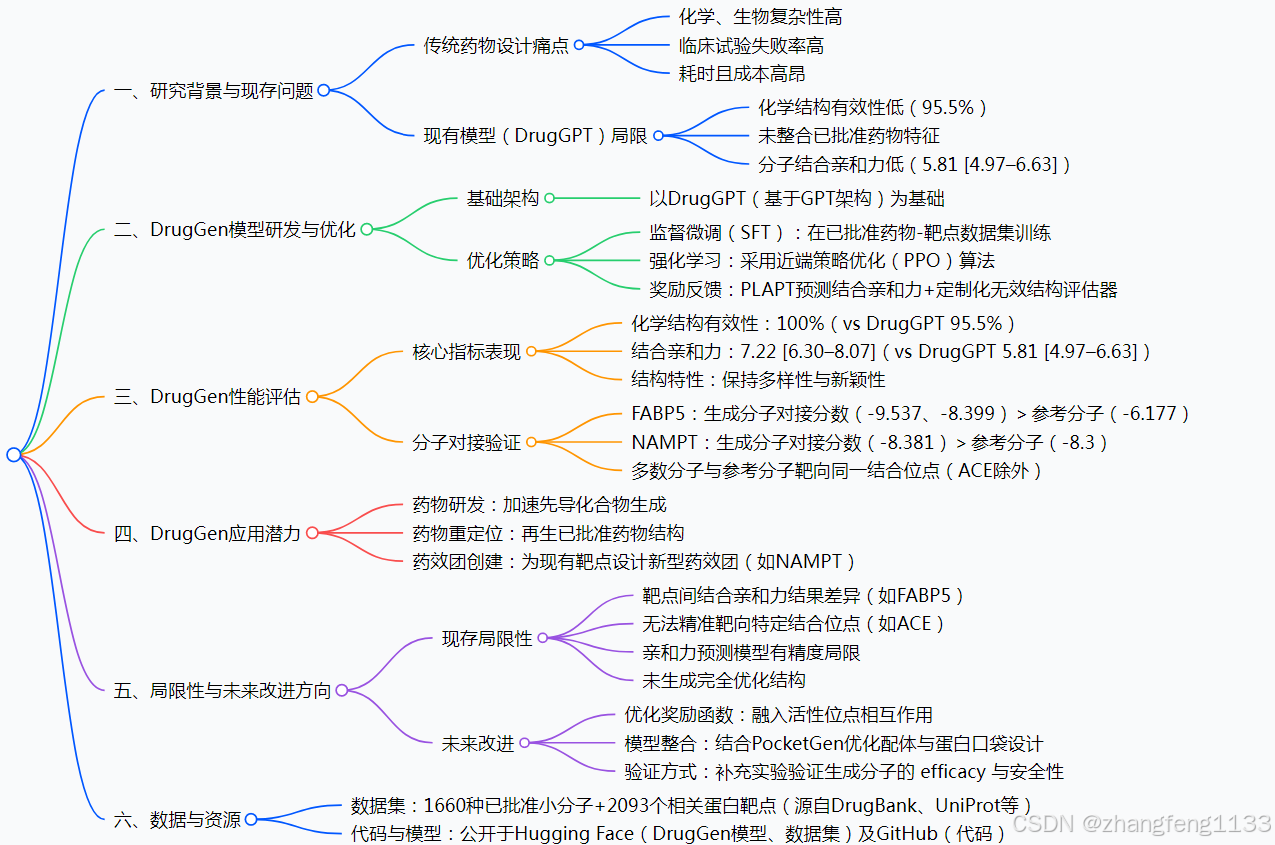
DrugGPT 提出时间与相关背景梳理
根据现有文档及补充搜索信息,DrugGPT 的提出时间需结合不同研发团队的技术成果与版本迭代历史综合判断,核心可追溯至 2023年,具体细节如下:
一、首次公开提出:2023年7月(西安交通大学团队)
最早明确提及“DrugGPT”模型并阐述其技术原理的公开信息,来自西安交通大学第一附属医院 Yuesen Li 团队发表于 2023年7月15日 的研究(摘要2)。该研究发表在预印本平台 BioRxiv 上,首次提出“基于自回归模型 GPT 的配体设计策略 DrugGPT”,核心目标是通过 GPT 架构探索化学空间、发现特定蛋白质的靶向配体,为药物研发提供新工具。这一时间点是目前可追溯的 DrugGPT 首次公开提出的关键节点,且明确了其在药物分子生成领域的初始定位。
此外,arXiv 上 2025年11月发布的论文(摘要5)也间接佐证了这一时间线,文中引用“DrugGPT (Li et al., 2023)”,进一步确认该模型在 2023年已有公开研究成果,与西安交通大学团队的 2023年7月工作形成呼应。
二、版本迭代与功能扩展:2024年(专项工具与验证阶段)
2023年提出后,DrugGPT 进入版本优化与功能扩展阶段,相关团队在 2024年发布了多个迭代版本并完成关键验证,具体时间线如下:
- 2024年3月31日:发布稳定版本“DrugGPT_v1.0”,同时规划创建“drug gpt_toolbox”(存储后处理工具脚本)和“druggpt_train”(存储训练脚本)两个仓库,明确模型核心定位为“候选药物分子生成”(摘要1);
- 2024年4月3日:升级至“DrugGPT_v1.1”,优化模型稳定性并新增 WebUI 交互功能,同时预告后续版本将加入“分子原子数量控制”功能(摘要1);
- 2024年5月16日:迭代至“DrugGPT_v1.2”,正式实现“原子数量控制”能力,但因兼容性问题移除此前新增的 WebUI(摘要1);
- 2024年7月30日:完成所有湿实验室(wet-lab)验证,证实 DrugGPT 具备“配体优化能力”,标志模型从理论设计走向实验验证阶段(摘要1);
- 2024年8月11日:新增“配体能量最小化”功能,推出专用工具“druggpt_min_multi.py”,优化多配体高效能量最小化处理效率(摘要1)。
三、其他团队相关模型:2024-2025年(同名场景化应用)
需注意的是,2024-2025年另有其他团队推出同名“DrugGPT”模型,但属于聚焦特定场景的衍生应用,与 2023年提出的“药物分子生成类 DrugGPT”并非同一模型,其公开时间不影响核心版本的“提出时间”判定:
- 2024年4月:牛津大学研发“DrugGPT”,以聊天机器人形式提供处方指导、药物相互作用提示等临床用药支持,核心应用场景为“用药安全辅助”,而非药物分子生成(摘要4);
- 2025年10月:西湖大学郑冶枫团队联合多方开发“DrugGPT”,定位为“基于知识协作机制的大语言模型药物分析系统”,侧重药物分析而非分子生成(摘要3)。
四、总结:DrugGPT 核心版本的提出时间
综合技术源头与公开文献,DrugGPT(聚焦药物分子生成、基于GPT自回归架构的核心版本)首次提出于2023年7月,由西安交通大学第一附属医院 Yuesen Li 团队在 BioRxiv 预印本中公开;2024年为该模型的“版本迭代与实验验证期”,逐步完善功能并完成湿实验室验证;2024-2025年其他团队的同名模型为场景化衍生应用,不改变核心版本的提出时间追溯。