【R语言】构建GO、KEGG相关不同物种的R包
构建GO、KEGG相关不同物种的R包
数据准备
- 物种对应的CDS基因组文件(NCBI可获得)
- KEGG中的ko0001.json
- tax_id = “10090”,这里以小鼠举例
genus = “Mus”
species = “musculus”
软件安装
eggnog-mapper
git clone https://github.com/jhcepas/eggnog-mapper.git
在http://eggnog5.embl.de/download/emapperdb-5.0.2/下下载这几个文件:
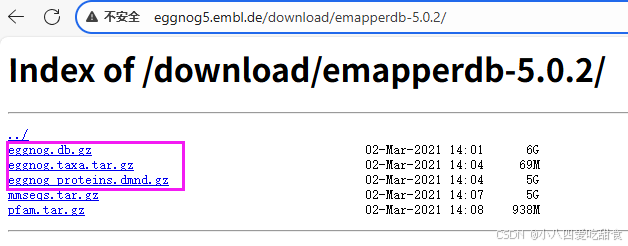
然后放到eggnog-mapper/data/下,并用gunzip *gz解压。
echo ‘export PATH=/public/user/software/eggnog-mapper:$PATH’ >> ~/.bashrc
source ~/.bashrc
emapper.py -i 物种.cds.fa -o 物种名称 -d euk --cpu 10 --dbmem --itype CDS
id=mm39 #刚才的前缀
sed '/^##/d' ${id}.emapper.annotations| sed 's/#//g'| awk -vFS="\t" -vOFS="\t" '{print $1,$9,$10,$12}' > ${id}.annotations
构建R包
#install.packages("")
library(dplyr)
library(stringr)
library(jsonlite)
library(AnnotationForge)
options(stringsAsFactors = F)
emapper <- read.table("mm39.annotations", header=TRUE, sep = "\t",quote = "")
#将空值替换为NA,方便后续使用na.omit()函数提出没有注释到的行
emapper[emapper==""]<-NA
# library(dplyr)
gene_info <- emapper %>% dplyr::select(GID = query, GENENAME = Preferred_name) %>% na.omit()
gos <- emapper %>% dplyr::select(query, GOs) %>% na.omit()gene2go = data.frame(GID = character(),GO = character(),EVIDENCE = character())
# library(stringr)
gos_list <- function(x){the_gos <- str_split(x[2], ",", simplify = FALSE)[[1]]df_temp <- data.frame(GID = rep(x[1], length(the_gos)),GO = the_gos,EVIDENCE = rep("IEA", length(the_gos)))return(df_temp)
}
gene2gol <- apply(as.matrix(gos),1,gos_list)
gene2gol_df <- do.call(rbind.data.frame, gene2gol)
gene2go <- gene2gol_df
gene2go$GO[gene2go$GO=="-"]<-NA
gene2go<-na.omit(gene2go)
gene2ko <- emapper %>% dplyr::select(GID = query, Ko = KEGG_ko)
gene2ko$Ko[gene2ko$Ko=="-"]<-NA
gene2ko<-na.omit(gene2ko)
gene2kol <- apply(as.matrix(gene2ko),1,gos_list)
gene2kol_df <- do.call(rbind.data.frame, gene2kol)
gene2ko <- gene2kol_df[,1:2]
colnames(gene2ko) <- c("GID","Ko")
gene2ko$Ko <- gsub("ko:","",gene2ko$Ko)
# library(jsonlite)
# 下面的json = "ko00001.json",如果你下载到其他地方,记得加上路径
update_kegg <- function(json = "ko00001.json") {pathway2name <- tibble(Pathway = character(), Name = character())ko2pathway <- tibble(Ko = character(), Pathway = character())kegg <- fromJSON(json)for (a in seq_along(kegg[["children"]][["children"]])) {A <- kegg[["children"]][["name"]][[a]]for (b in seq_along(kegg[["children"]][["children"]][[a]][["children"]])) {B <- kegg[["children"]][["children"]][[a]][["name"]][[b]] for (c in seq_along(kegg[["children"]][["children"]][[a]][["children"]][[b]][["children"]])) {pathway_info <- kegg[["children"]][["children"]][[a]][["children"]][[b]][["name"]][[c]]pathway_id <- str_match(pathway_info, "ko[0-9]{5}")[1]pathway_name <- str_replace(pathway_info, " \\[PATH:ko[0-9]{5}\\]", "") %>% str_replace("[0-9]{5} ", "")pathway2name <- rbind(pathway2name, tibble(Pathway = pathway_id, Name = pathway_name))kos_info <- kegg[["children"]][["children"]][[a]][["children"]][[b]][["children"]][[c]][["name"]]kos <- str_match(kos_info, "K[0-9]*")[,1]ko2pathway <- rbind(ko2pathway, tibble(Ko = kos, Pathway = rep(pathway_id, length(kos))))}}}save(pathway2name, ko2pathway, file = "kegg_info.RData")}
# 调用函数后在本地创建kegg_info.RData文件,以后只需要载入 "kegg_info.RData"即可
update_kegg()
# 载入kegg_info.RData文件
load(file = "kegg_info.RData")
gene2pathway <- gene2ko %>% left_join(ko2pathway, by = "Ko") %>% dplyr::select(GID, Pathway) %>% na.omit()
tax_id = "10090"
genus = "Mus"
species = "musculus"
# gene2go <- unique(gene2go)
# gene2go <- gene2go[!duplicated(gene2go),]
# gene2ko <- gene2ko[!duplicated(gene2ko),]
# gene2pathway <- gene2pathway[!duplicated(gene2pathway),]
# gene_info <- gene_info[!duplicated(gene_info),]
makeOrgPackage(gene_info=gene_info,go=gene2go,ko=gene2ko,pathway=gene2pathway,version="1.34.1", #版本,使用?makeOrgPackage,拉到最下面查看maintainer = "your name <邮箱>", #修改为你的名字和邮箱author = "your name <邮箱>", #修改为你的名字和邮箱outputDir = ".", #输出文件位置tax_id=tax_id, #你在NCBI上查并定义的idgenus=genus, #你在NCBI上查并定义的属名species=species, #你在NCBI上查并定义的种名goTable="go")R CMD build org.Mmusculus.eg.db
获得org.Mmusculus.eg.db.tar.gz,
install.packages("org.Mmusculus.eg.db.tar.gz", repos=NULL)
library(org.Mmusculus.eg.db)