如何批量获取蛋白质序列的所有结构域(domain)数据-1
文章目录
- 背景:
- 一,综合性蛋白质数据库的API接口:Uniprot为例
- 总结:
- 重点:如何使用&接口访问
- 1,Retrieving individual entries 检索单个条目
- 2,Retrieving entries via queries 通过query查询检索条目
- (1)Formats 格式
- (2)有哪些格式可用?
- (3)如何构建搜索url?
- 1️⃣高级搜索
- 2️⃣如何构建高级搜索?
- (1)文本搜索:核心在于查询语法(query syntax)
- (2)文本搜索的对象:核心在于查询字段(query field)
- (3)返回字段:return fields
- (4)Python示例—在 Python 脚本中立即使用搜索结果
- (1)结果数量较少:使用流stream
- (2)结果数量较多:使用分页pagination
- (5)Python示例—将搜索结果保存到磁盘
- (1)结果数量较少,使用流stream
- (2)结果数量较多,使用分页pagination
- 3,Query/Return fields 查询/返回字段
- 4,其他
- (1)不同数据库之间的ID映射(mapping)
- (2)REST API 中的 HTTP 状态码
- 5,两个简单的脚本示例
- (1)没有蛋白质id,想找到符合query条件的蛋白质id(及其数据)
- (2)有了蛋白质id,想获取对应蛋白质的某些属性数据
- (3)一些补充:
对于一条蛋白质序列,如果有名有姓,也就是有ID,比如说uniprot ID,比如说CTCF human蛋白,uniprot ID就是P49711,那么其实就可以到各种结构域数据库中直接搜索,因为现有的蛋白质其实大多数结构域、功能关系都已经非常明了,解析出来了,可以参考的数据库也非常多,本质就是爬虫。
如果我们只有序列数据,也就是无名无姓,可能是某个已知蛋白质的某种变体、isoform,或者蛋白质组学(比如说质谱)等实验中获取的某个全新的肽段、蛋白质片段,那么这个时候想要了解这条蛋白质序列的结构域,就是预测问题,本质就是序列分析(生物信息)。因为结构域分割以及注释直到现在依旧是一个生信benchmark问题,SOTA。旧model是基于序列、基于序列比对/进化,近几年大多是基于结构的深度学习model。
背景:
我有很多蛋白质,但是只有纯序列数据(也就是fasta),我不确定这些蛋白质的id是什么,我只想系统性地、统一地为这些蛋白质,从序列角度去注释结构域。
如果是已知蛋白质,就是数据库查询(query&reference)问题;
如果是未知蛋白质,那就是结构域预测/注释问题,比如说Blast或者是HMM系列的,可以参考之前的博客:https://blog.csdn.net/weixin_62528784/article/details/151230484?spm=1001.2014.3001.5502。
总之问题就是:我想通过程序,快速地获取手头上一大堆蛋白质序列的结构域注释,并且尽可能地要求结构域注释信息可靠。
综合性蛋白质数据库,个人接触的主要是Uniprot、InterPro
一,综合性蛋白质数据库的API接口:Uniprot为例
这种情况,比如符合已知蛋白质“信息”(就是有名有姓),比如说Uniprot数据库,
我们都知道,Uniprot数据库并不是单纯存储蛋白质结构域的数据库,它其实是一个综合性数据库,
只不过每一个蛋白质信息条目中,会援引到结构域数据库的注释条目。
还是以CTCF human为例子:

我知道这个蛋白质的gene、organism等基本信息,
其实我就可以直接用统一地ID信息去提取这些蛋白质在这些数据库中的结构域domain数据。
比如说CTCF human就有Zinc Finger Domain
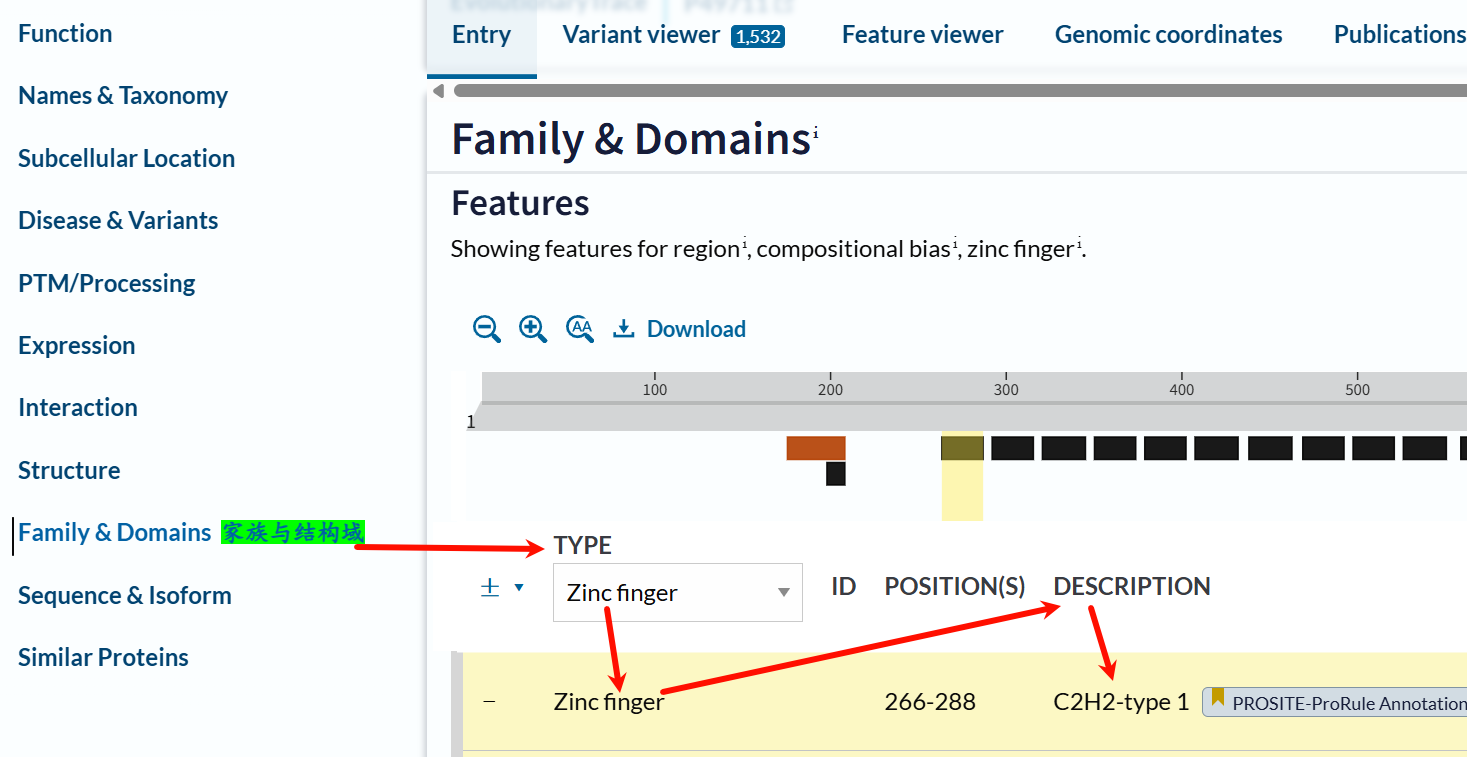
以上只是一unprot注释为例,
实际上我最后一个箭头所指的地方,其实就是PROSITE数据库注释,也就是uniprot此处的注释也是援引其他子数据库的。
这里其实就有一个问题,如果是你来做,比如说导师给你分配了个任务,收集这个蛋白质的所有结构域数据,你是直接到uniprot这个条目里爬虫爬一下就OK了吗?
其实你并不能确保uniprot中援引的注释数据库是否全面,
比如说这里为什么用PROSITE数据库的注释?
其实只要你往下再拉一点:
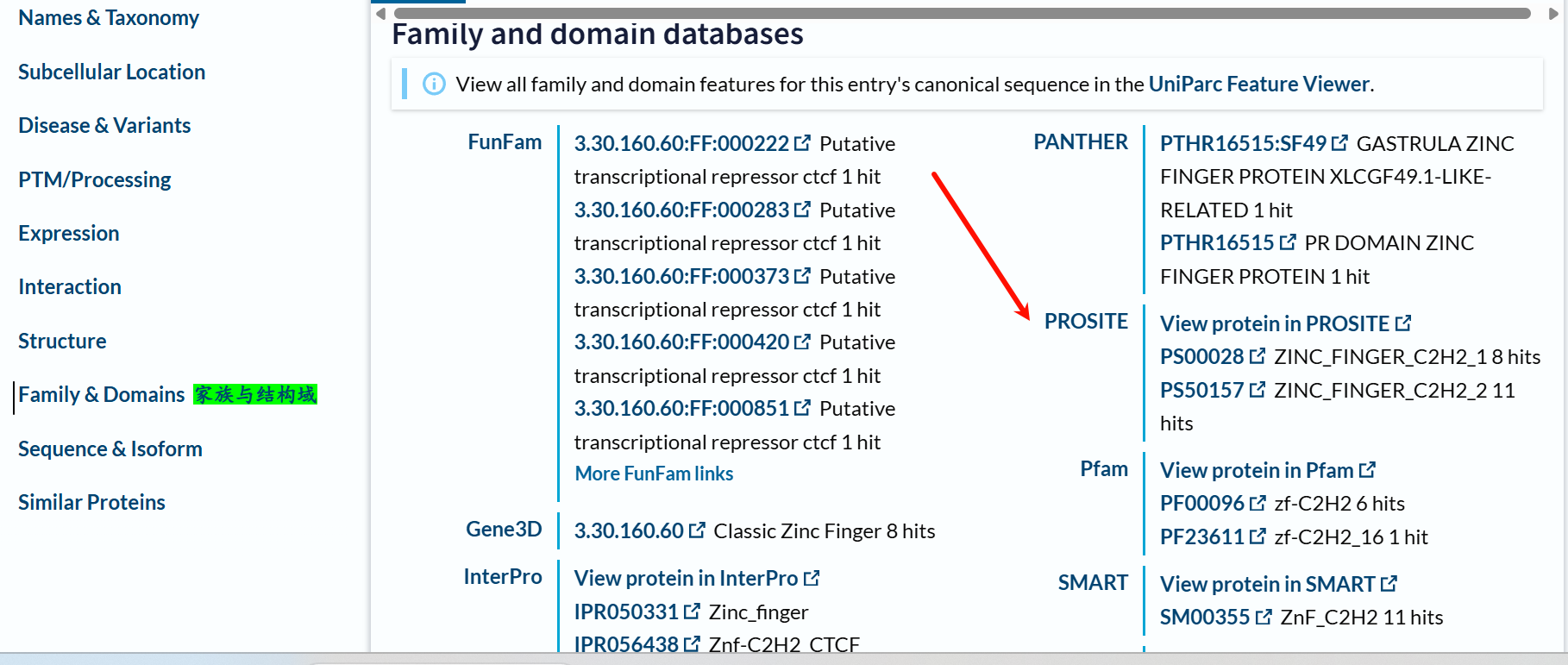
就会发现有更多的结构域数据库,
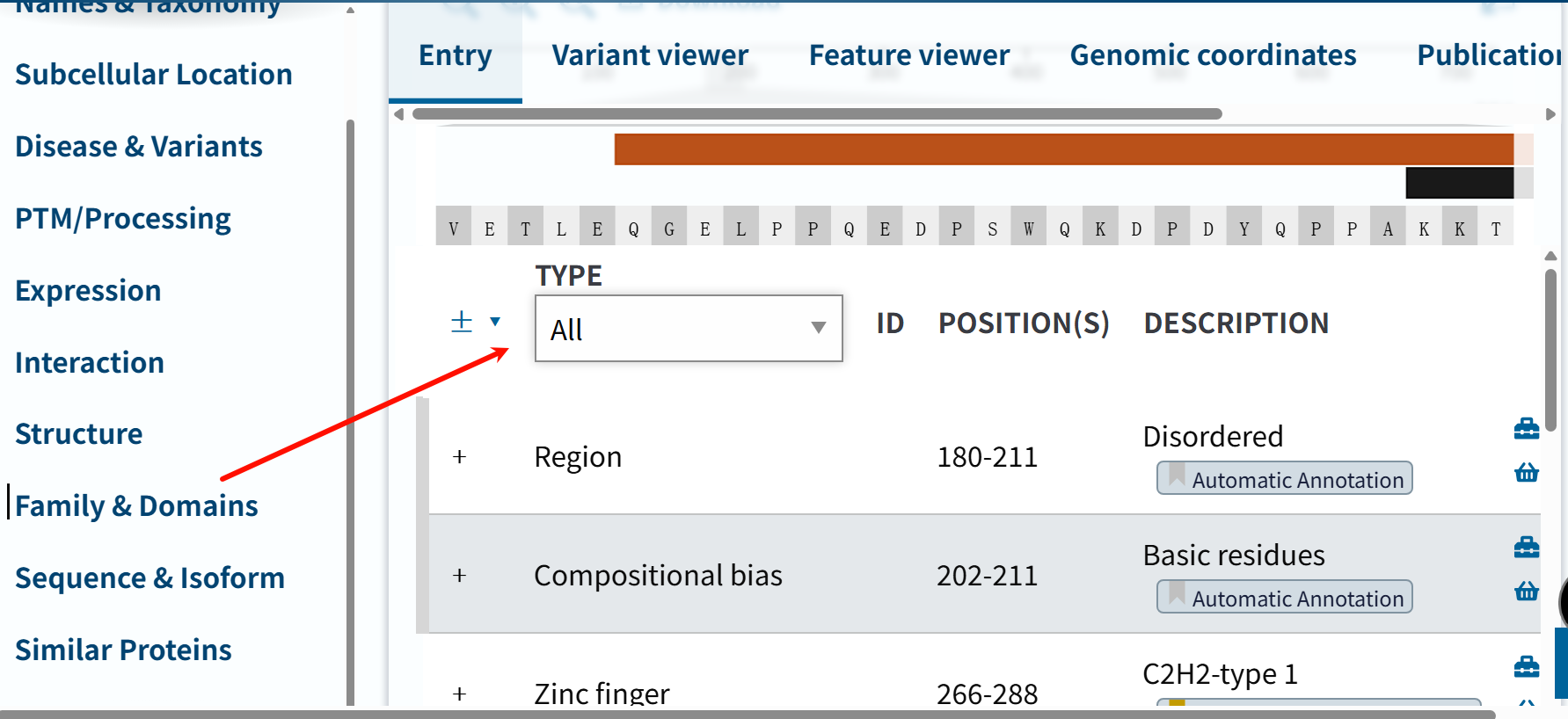
我箭头所指的就是PROSITE,这里是11 hits,也就是我们一般研究human CTCF蛋白公认的结构域组成。
问题就是:
为什么一定要用PROSITE呢?uniprot数据库也可以用其他的,比如说Pfam,从图中我们可以看到,大概就只有6或1 hit。对于其他的蛋白质,如果是我们不了解的,你怎么能够放心地直接爬虫uniprot的数据呢,
万一对于蛋白质A,uniprot用了结构域数据库B的数据,对于蛋白质C,又用了结构域数据库D的数据,这些都是不可控的。
本质就是注释工具太多了、注释规则太复杂,只是看uniprot汇总的注释,总感觉注释质量令人担忧。

https://www.uniprot.org/uniparc/UPI0000000DDE/feature-viewer
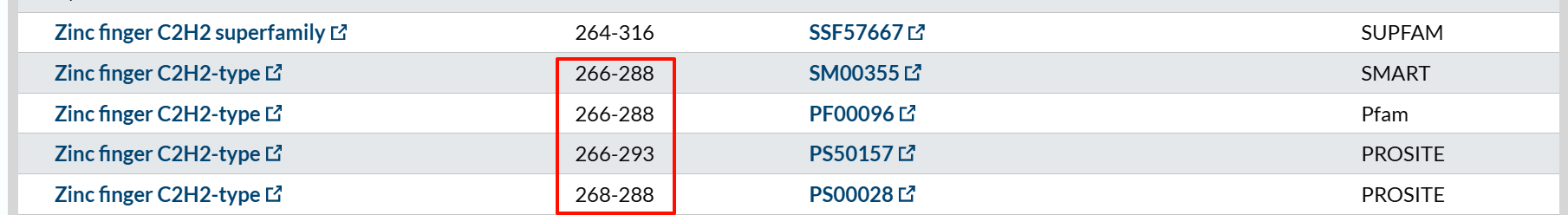
如上图红方框所示,就是单单1个结构域的注释就不统一,这是细节控所不能忍受的。
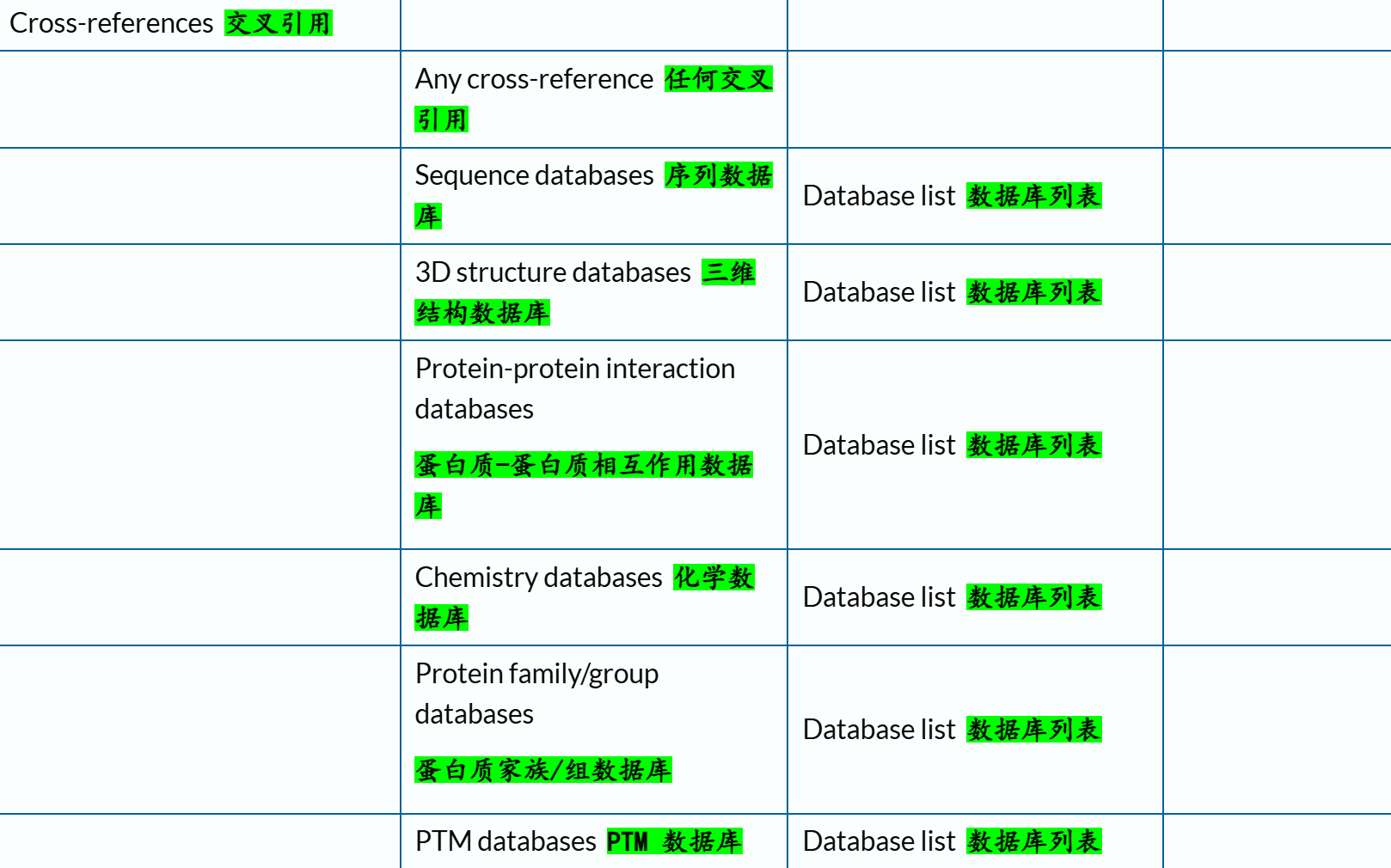
我们可以先来提纲挈领地查看一下Uniprot中所交叉参考的数据库信息:
链接参考:https://www.uniprot.org/database?facets=category_exact%3AFamily%20and%20domain%20databases&query=%2A

总结:
数据库来源复杂,虽然全面但是杂,而且注释条目不能确保质量(我指的是无法确认每一个蛋白质的注释来源)。
重点:如何使用&接口访问
至于如何使用程序接口来访问这些数据,也就是如何写代码快速获取注释数据,是我们工程的重点,也是本篇博客的重点。
官方API应用程序接口,链接参考:https://www.uniprot.org/help/programmatic_access
帮助文档链接:https://www.uniprot.org/help/api

1,Retrieving individual entries 检索单个条目
官方文档链接参考:https://www.uniprot.org/help/api_retrieve_entries
我们先来详细展示一下上述链接的效果:
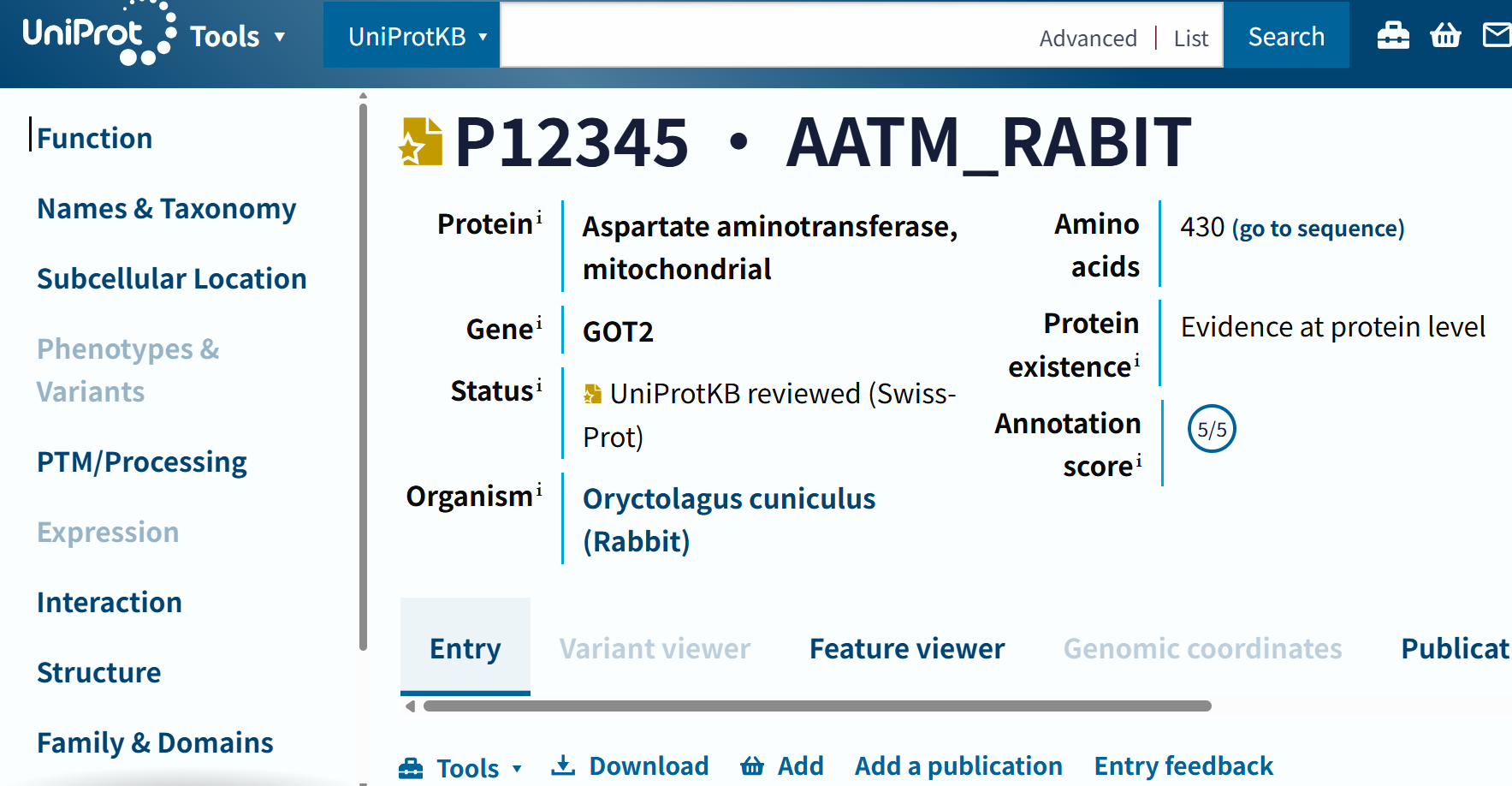
https://www.uniprot.org/uniprotkb/P12345
返回的直接就是对应ID条目的网页:是一个线粒体天冬氨酸转氨酶
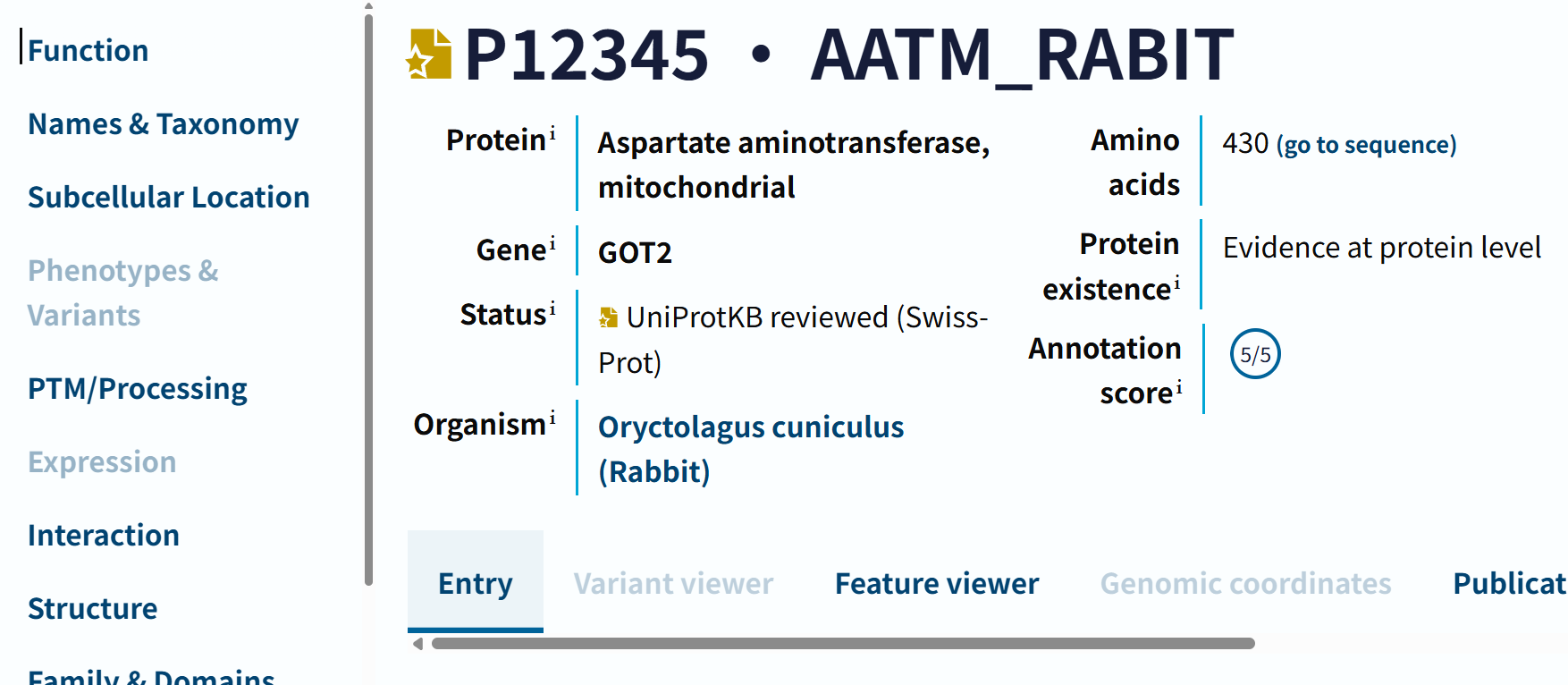
https://rest.uniprot.org/uniprotkb/P12345.txt
返回的是txt格式的网页:基本的数据是一致的,但是往下拉发现其实没有上面uniprot那个网页数据显示全
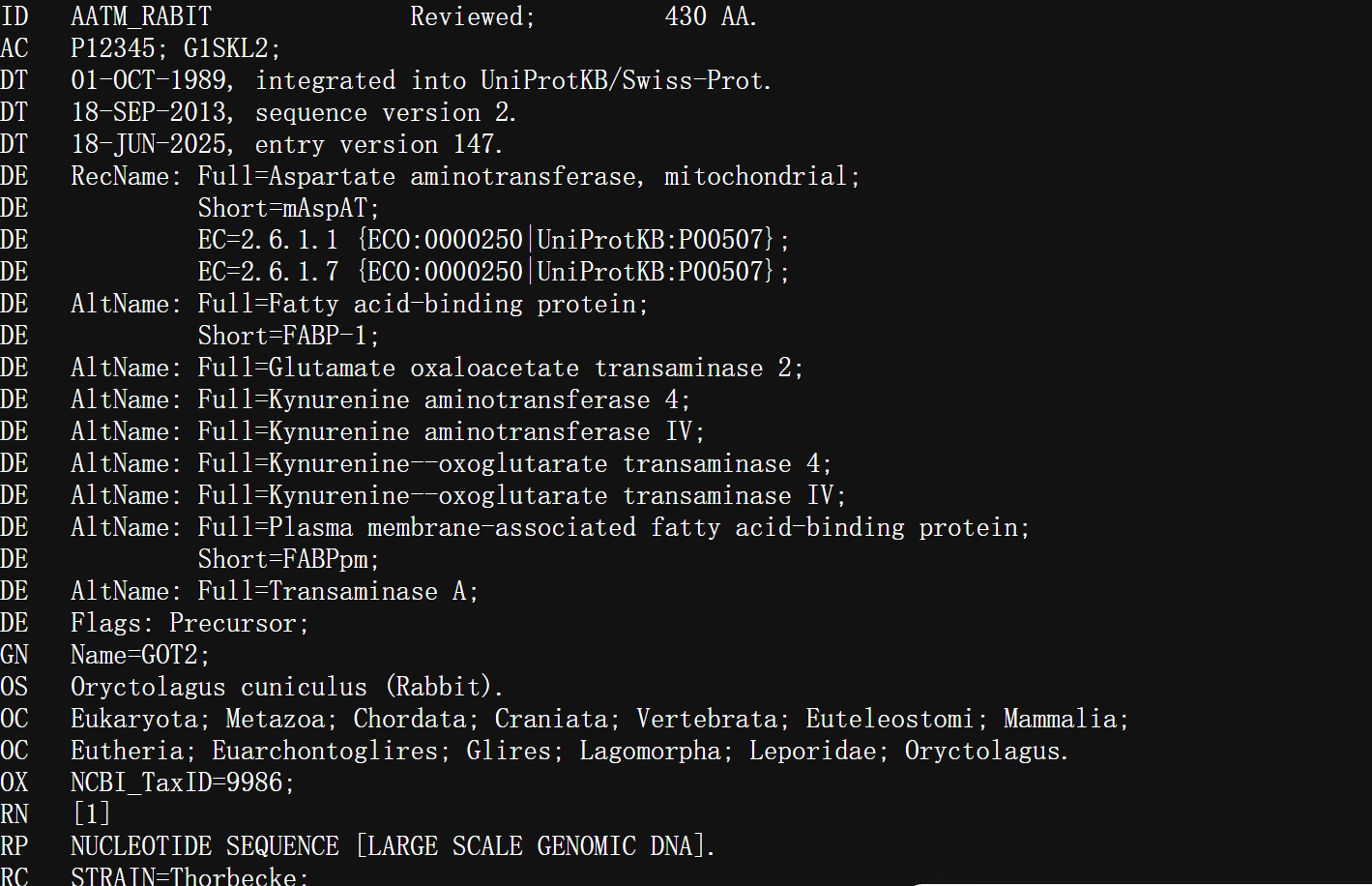
https://rest.uniprot.org/uniprotkb/P12345.xml
XML格式的数据结构化表示:
https://rest.uniprot.org/uniprotkb/P12345.rdf
rdf格式数据,

记事本中打开格式如下:


https://rest.uniprot.org/uniprotkb/P12345.rdf?include=yes

https://rest.uniprot.org/uniprotkb/P12345.fasta
就是该条目序列数据:

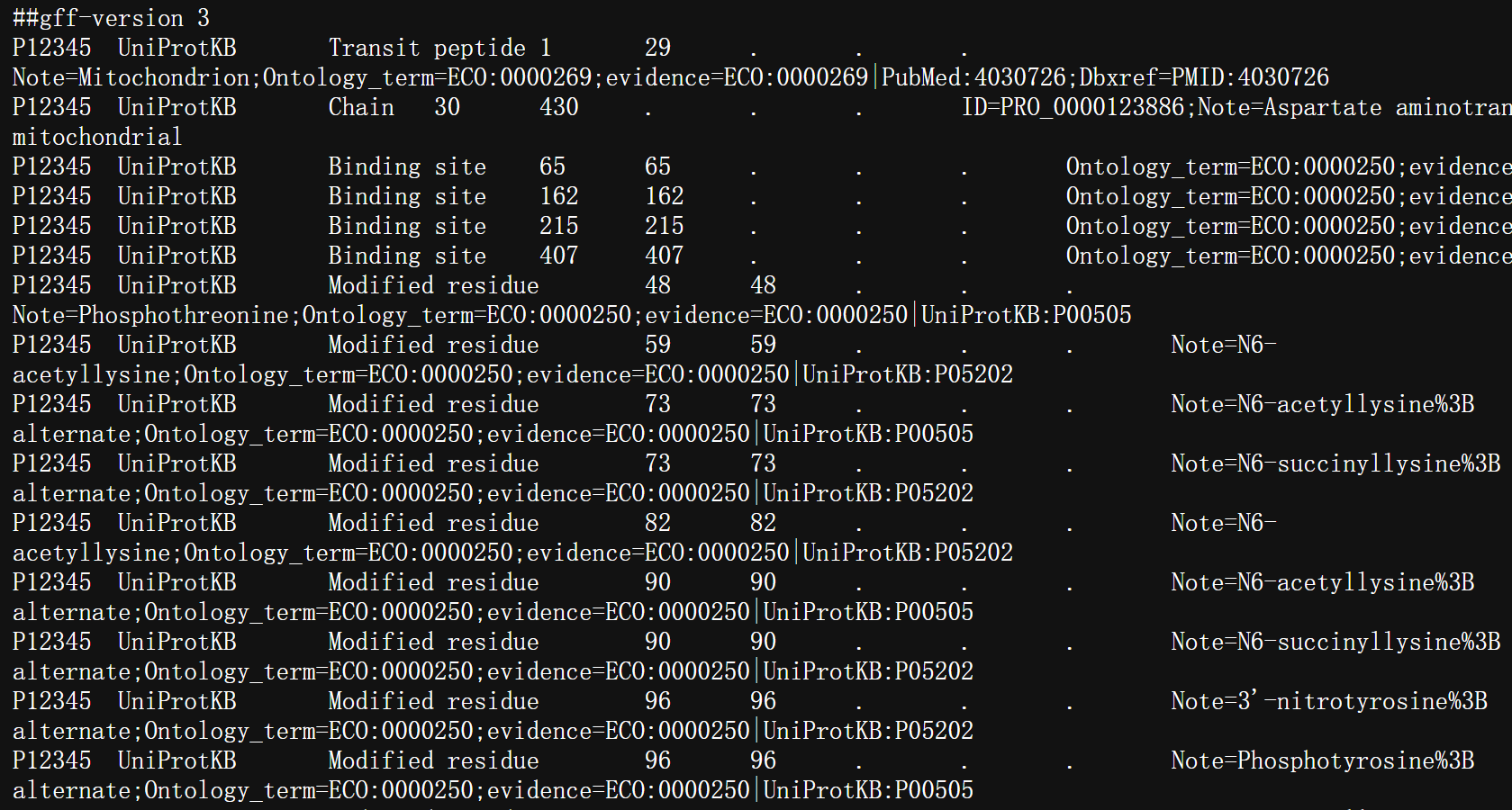
https://rest.uniprot.org/uniprotkb/P12345.gff
基本上只记录了修饰位点、功能位点数据
另外注意:

接口访问稳定性的问题:
参考链接:https://www.uniprot.org/help/linking_to_uniprot


官方建议在访问数据条目链接时,尽量使用accession number(AC序列号),而不是entry name,
两者区别参考:https://www.uniprot.org/help/difference_accession_entryname
entry name参考https://www.uniprot.org/help/entry_name
accession number参考https://www.uniprot.org/help/accession_numbers
简单来说:标题中的两者就分别对应AC与Entry_name,
很多人编程习惯用entry name来作为id展示,这其实是一个不好的习惯,
因为官方更建议使用AC,能够稳定、无偏地指向对应数据条目。


这里需要注意的是尽量使用Uniprot标识符来稳定访问,其他的访问并不一定稳定,比如说uniref,
Uniref的核心是“序列聚类”(将相似蛋白序列归为一类),而 UniProt 每次更新数据时,会重新计算这些聚类(比如纳入新发现的蛋白序列、优化聚类算法)。聚类结果变化后,每类对应的 “代表蛋白”(用来标识该聚类的核心蛋白)可能更换,进而导致原标识符对应的实际蛋白群体或核心信息改变。
所以用户若需稳定链接 UniProt 条目,可参考 “如何链接到 UniProt 条目”(https://www.uniprot.org/help/linking_to_uniprot) 的相关内容,UniRef 标识符不适合长期固定引用,需通过其他更稳定的方式关联 UniProt 数据” 。
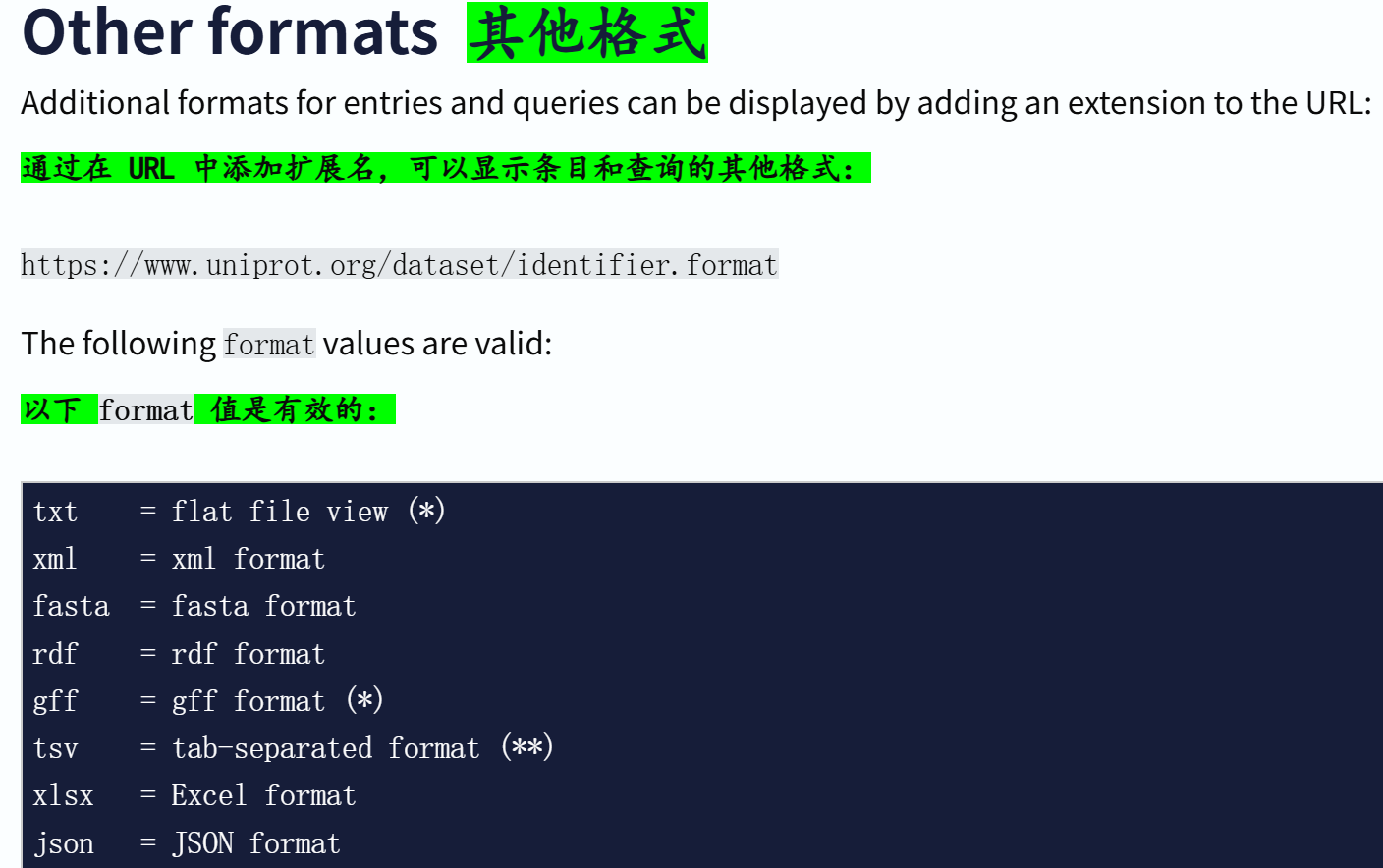
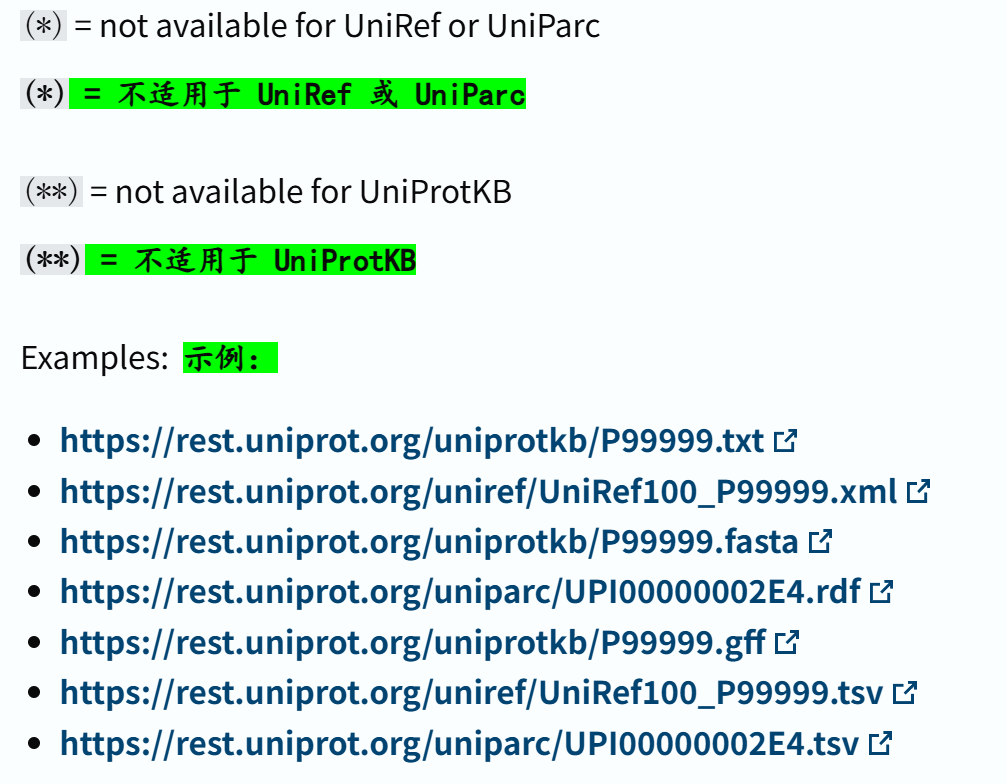
格式拓展名,就是前面展示的几个链接,效果也都见过:

我们再回到前面哪个rdf格式链接
https://rest.uniprot.org/uniprotkb/P12345.rdf?include=yes
可能会返回下列状态码status code:


http://purl.uniprot.org/uniprot/P12345
这个地址请求返回的也是纯网页,
2,Retrieving entries via queries 通过query查询检索条目
官方指南参考链接:https://www.uniprot.org/help/api_queries

简单解释一下这里的这两个链接:
虽然看上去有点不一样
https://www.uniprot.org/uniprotkb?query=(reviewed:true)%20AND%20(organism_id:9606)
UniProt 网站的交互式搜索链接,主要用于通过网页界面直观查看搜索结果。用户可以直接在浏览器中打开该链接,通过可视化页面浏览 “所有经过审核的人类项目”(即 reviewed:true 且 organism_id:9606 的 UniProtKB 条目),还能利用网页上的 “Download”“Customize data” 等功能进一步操作结果,属于面向普通用户的 “前端浏览工具”。
https://rest.uniprot.org/uniprotkb/search?query=(reviewed:true)%20AND%20(organism_id:9606)
UniProt REST API 的搜索请求链接,用于通过编程方式(如 Python 脚本、curl 命令)获取数据。它是网站数据的 “后端数据源”,返回的结果更适合机器解析(而非人工浏览),支持通过添加参数(如 format 指定数据格式、size 指定结果数量)自定义数据,还能结合分页、流式传输等方式批量获取数据,属于面向开发者的 “数据接口工具”。
简单来说,前者是网站链接,后者为API链接,
两个链接的本质关系是:网站链接(前者)通过调用 API 链接(后者)的后端能力,为用户提供可视化的前端浏览体验;而 API 链接则直接提供原始数据接口,方便用户通过代码批量处理或集成数据。
当然,我们的需求是个性化的,所以重点是如何自定义检索到的数据,以及如何分页浏览 API 所暴露的所有数据页。
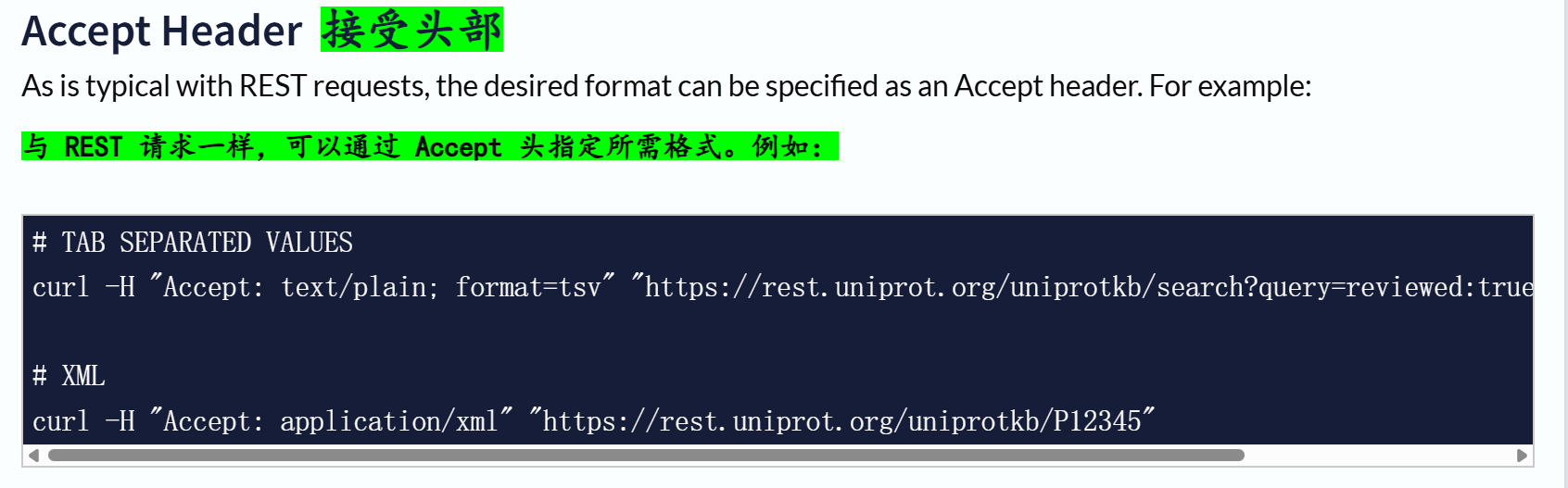
(1)Formats 格式

# TAB SEPARATED VALUES
"https://rest.uniprot.org/uniprotkb/search?query=reviewed:true+AND+organism_id:9606&format=tsv"# XML
"https://rest.uniprot.org/uniprotkb/P12345?format=xml"
# TAB SEPARATED VALUES
curl -H "Accept: text/plain; format=tsv" "https://rest.uniprot.org/uniprotkb/search?query=reviewed:true+AND+organism_id:9606"# XML
curl -H "Accept: application/xml" "https://rest.uniprot.org/uniprotkb/P12345"
和前面的格式相互对应。
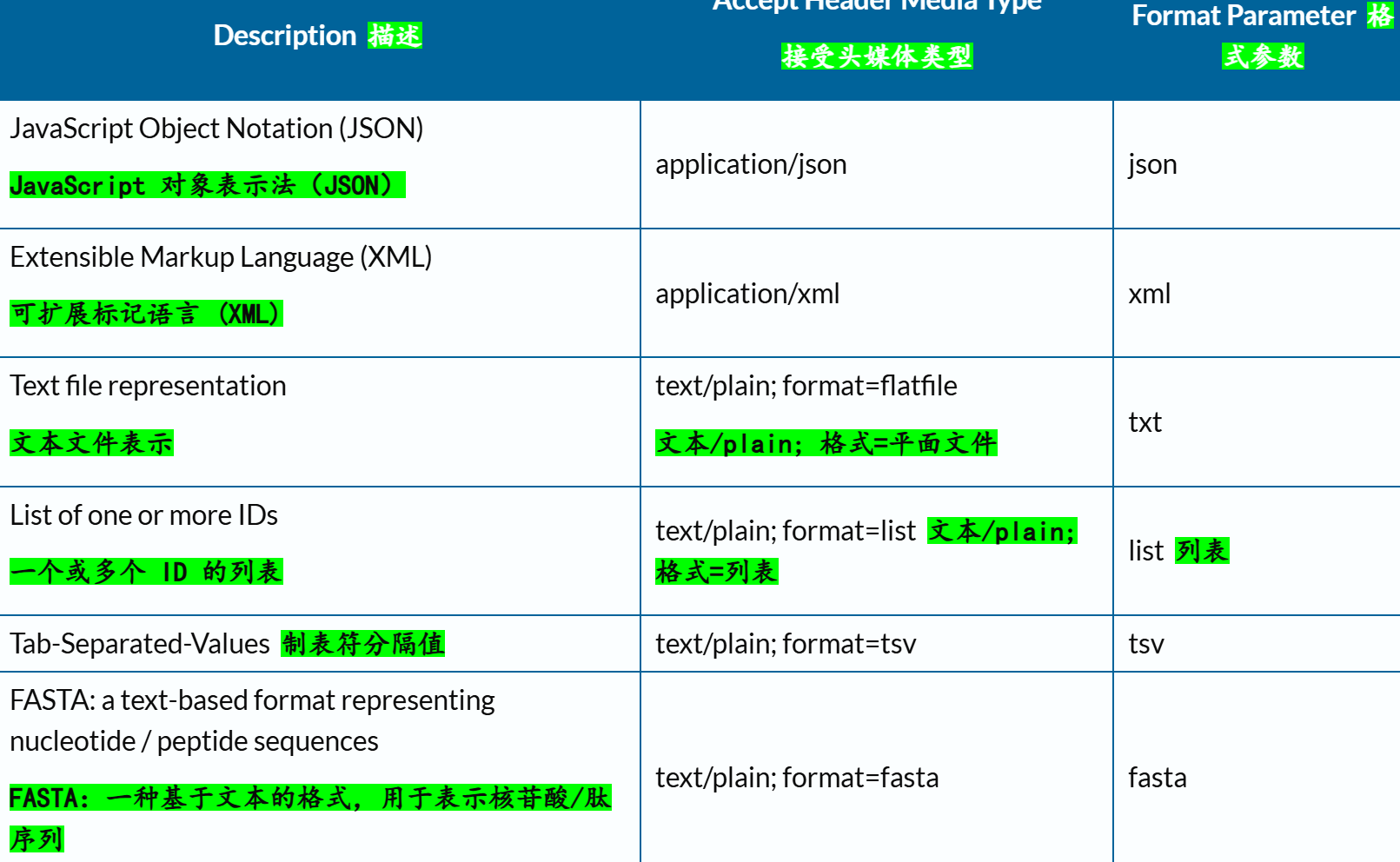
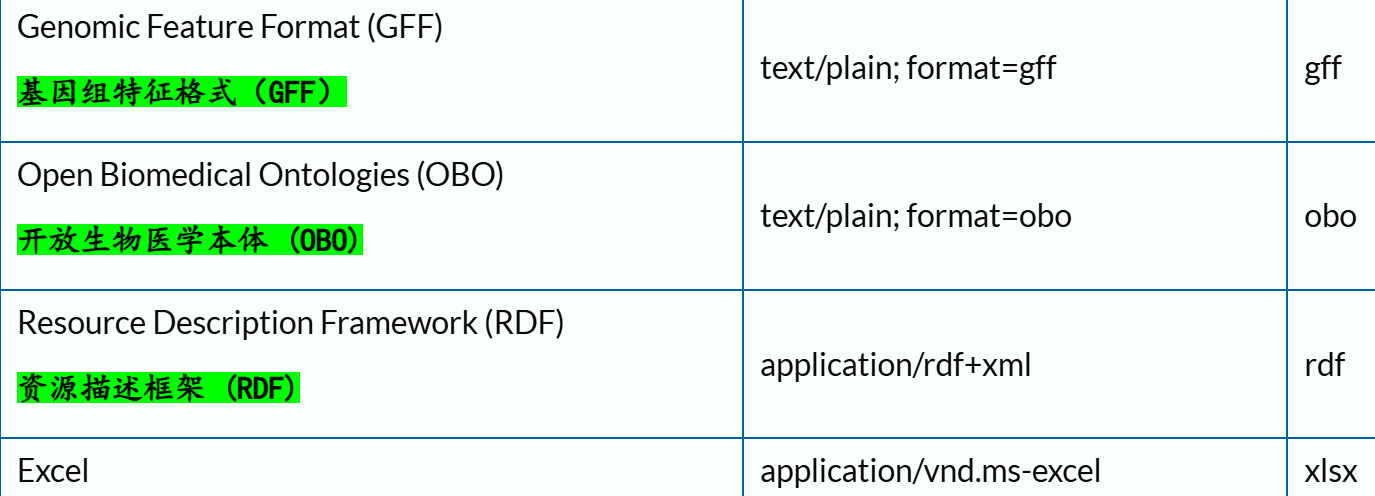
(2)有哪些格式可用?
通常来说,JSON和tsv格式是通用的。
示例:
## Accept header
curl -H "Accept: text/plain; format=flatfile" "https://rest.uniprot.org/uniprotkb/P12345"## Format Parameter
curl "https://rest.uniprot.org/uniprotkb/search?query=human&format=gff"
你要检索的item?format=
(3)如何构建搜索url?

1️⃣高级搜索
官方链接参考:https://www.uniprot.org/help/advanced_search


右上角这里点开之后,具体是:
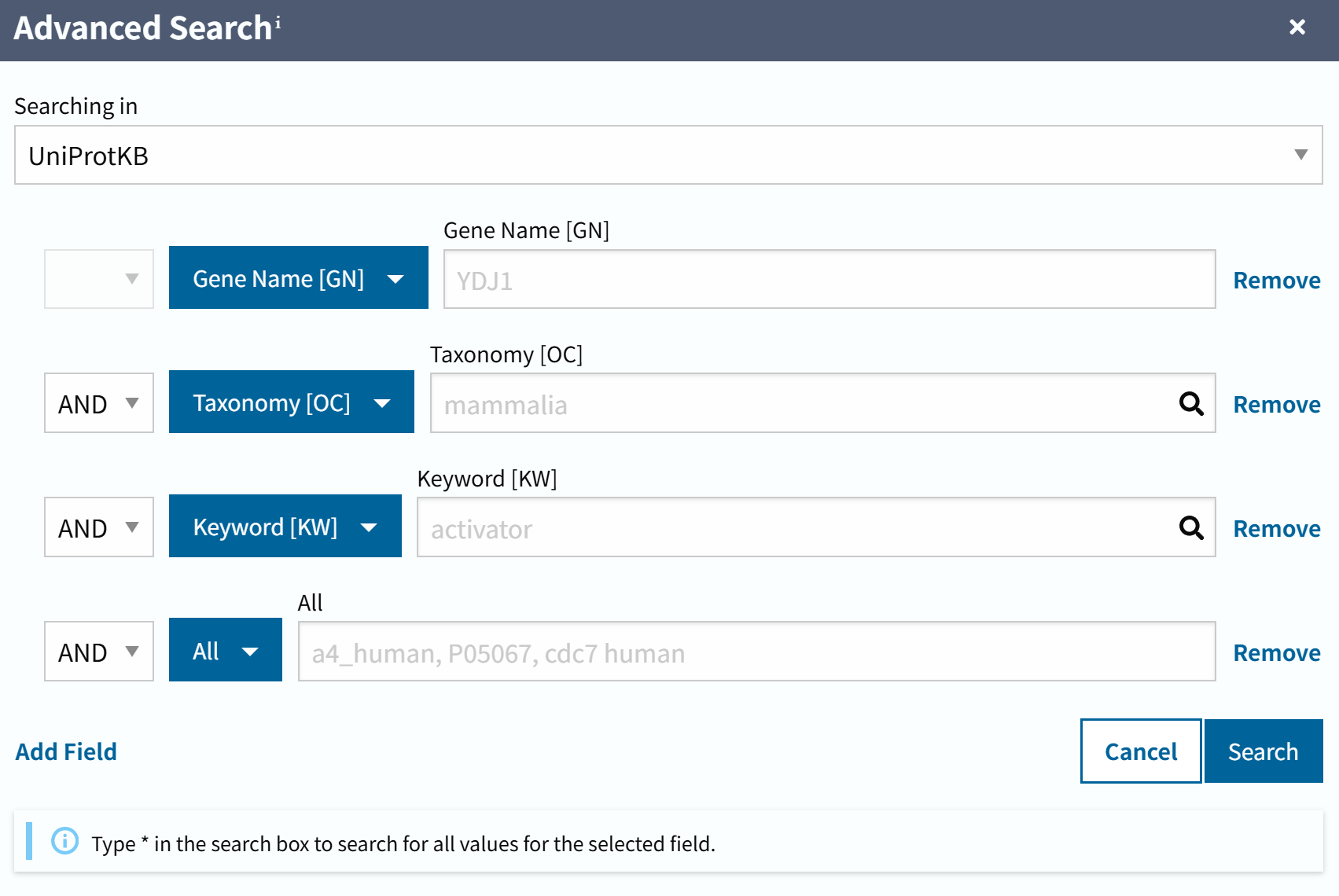
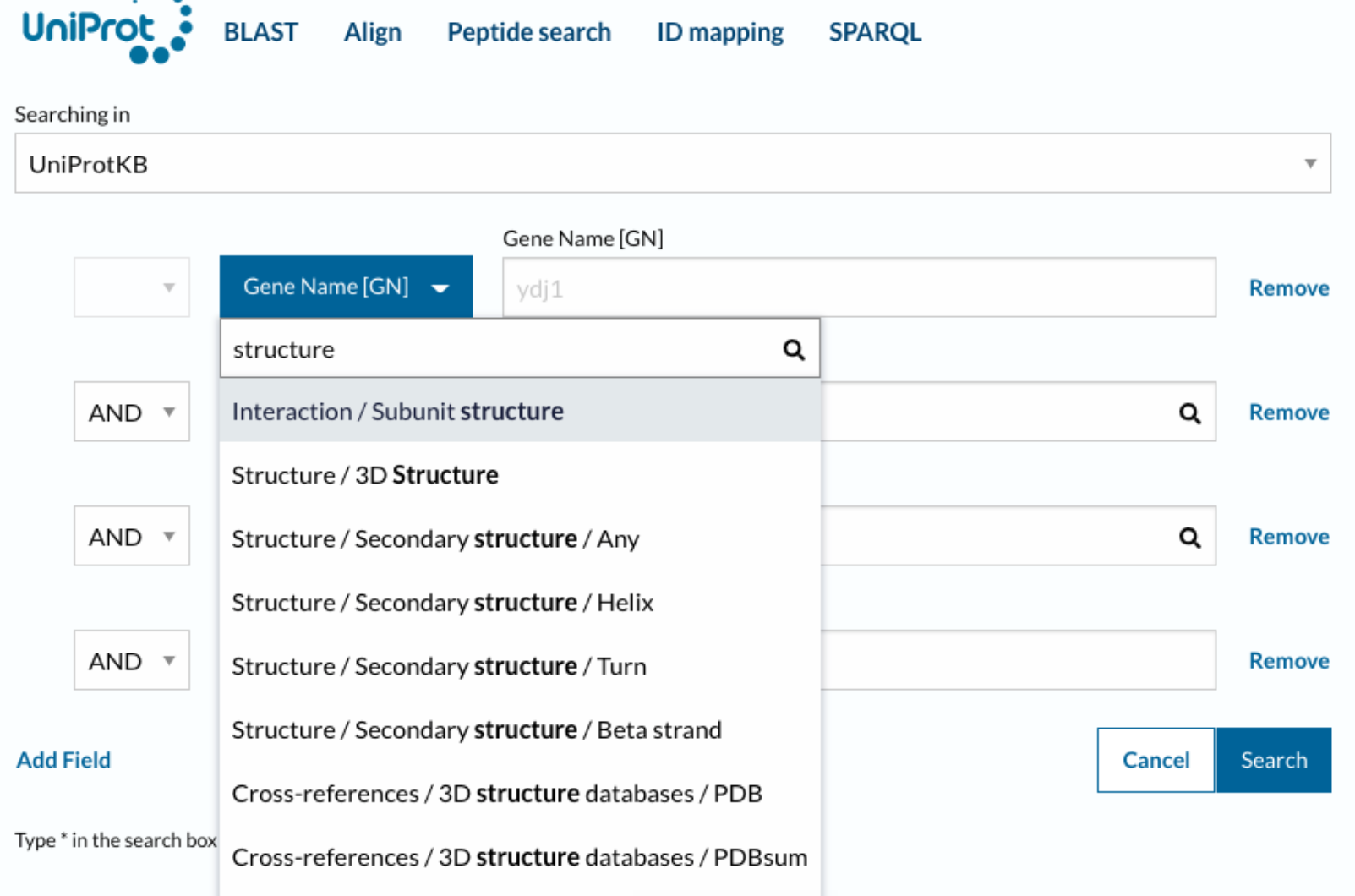
上面是图形化的界面,对应如何在程序中调用:



其中不少数据其实完全可以用于我们深度学习model的训练,
比如说上面条目中的:
功能(一般不太好量化,但是其中的各种位点数据,比较好量化,也容易放到model中进行训练之类),
蛋白质互作(其实就是蛋白质互作网络,可以抽象成图,GNN相关的数据输入),
结构(这个数据就不用说了,只要是涉及到分子机制的,底层要挖就得往结构上面去挖),
PTM翻译后修饰,其实这个和功能/位点那一块有点重;
序列(这个也不用说,最基础的feature,在LM中可以考虑token),
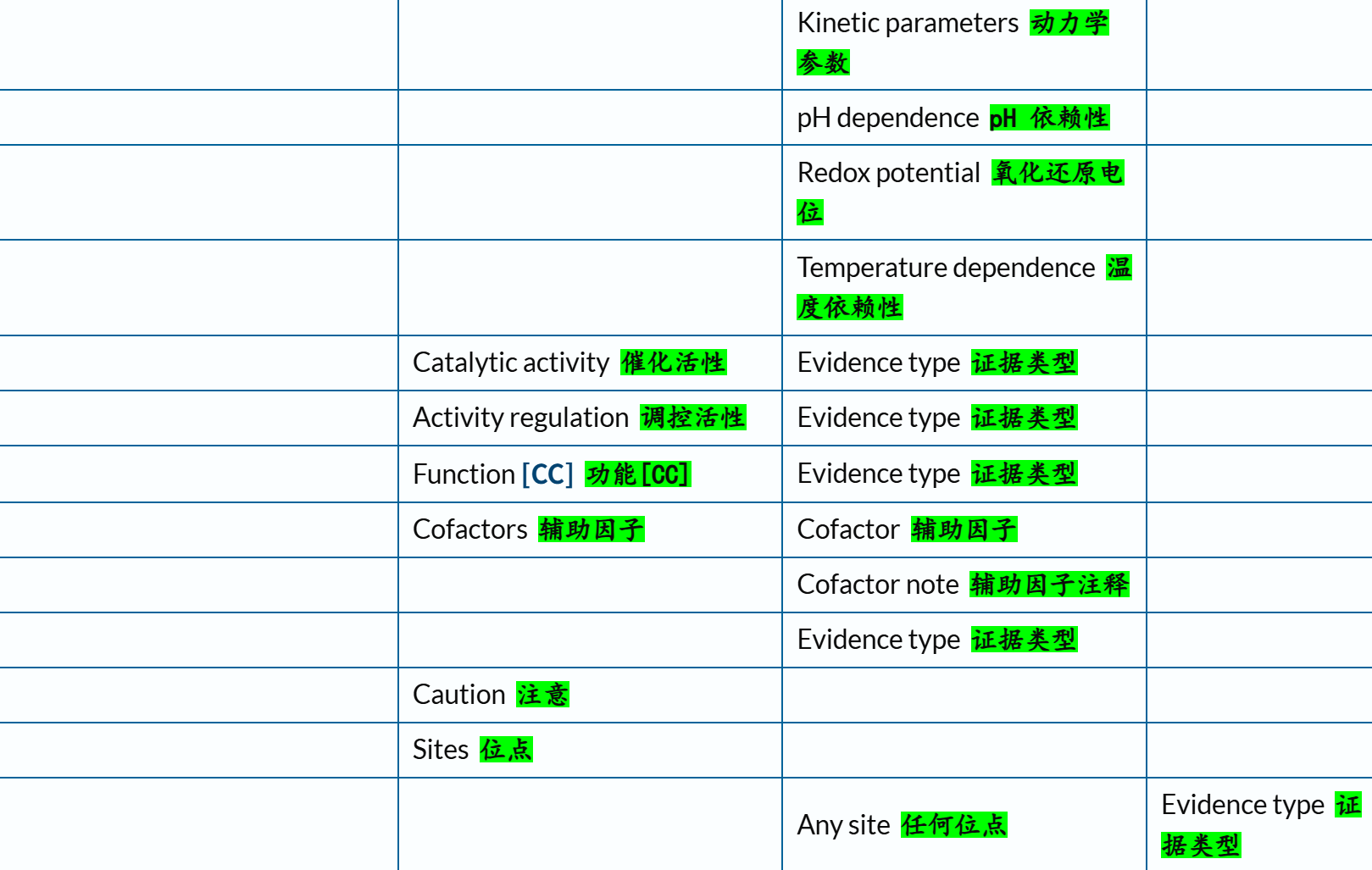
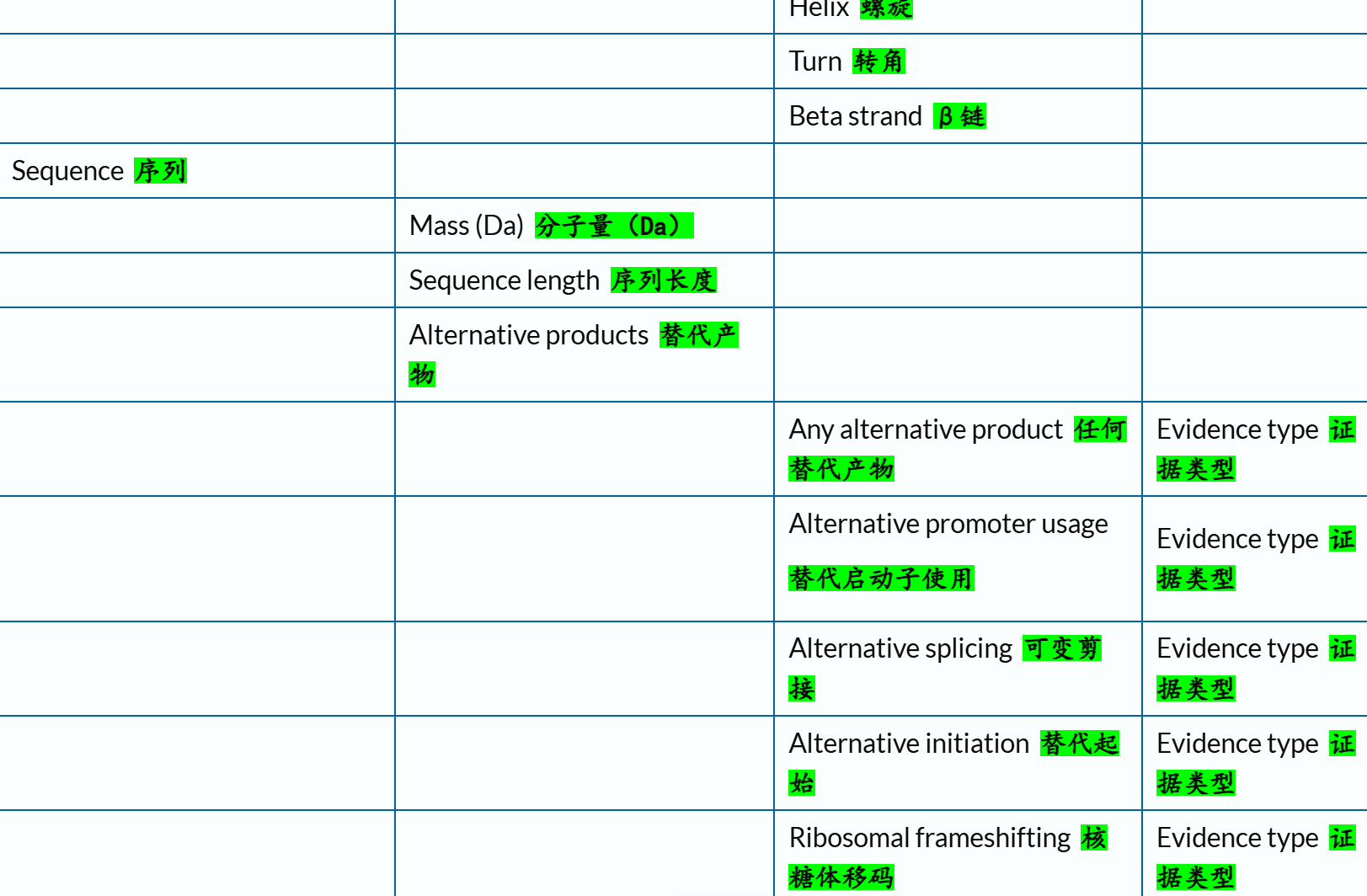
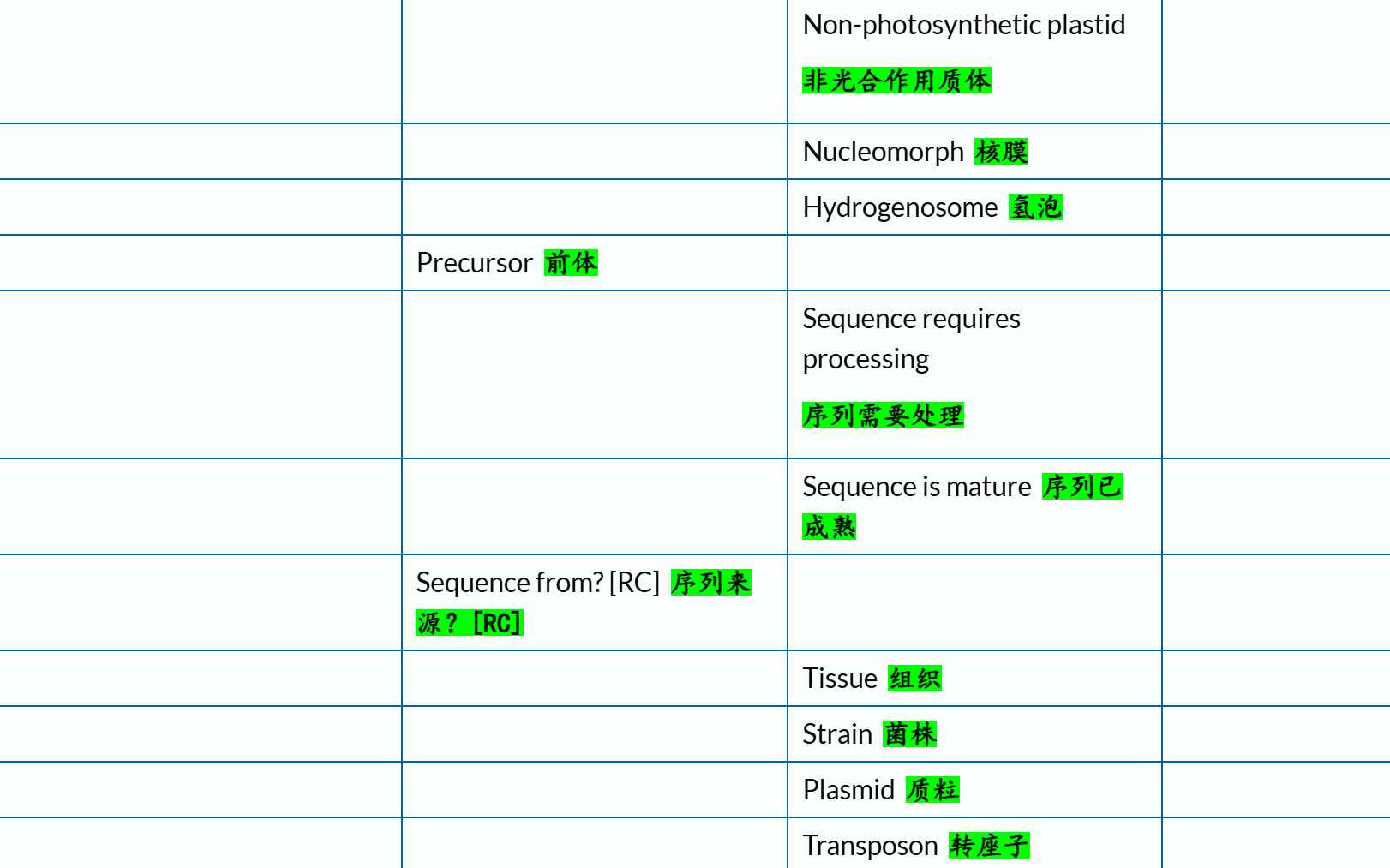
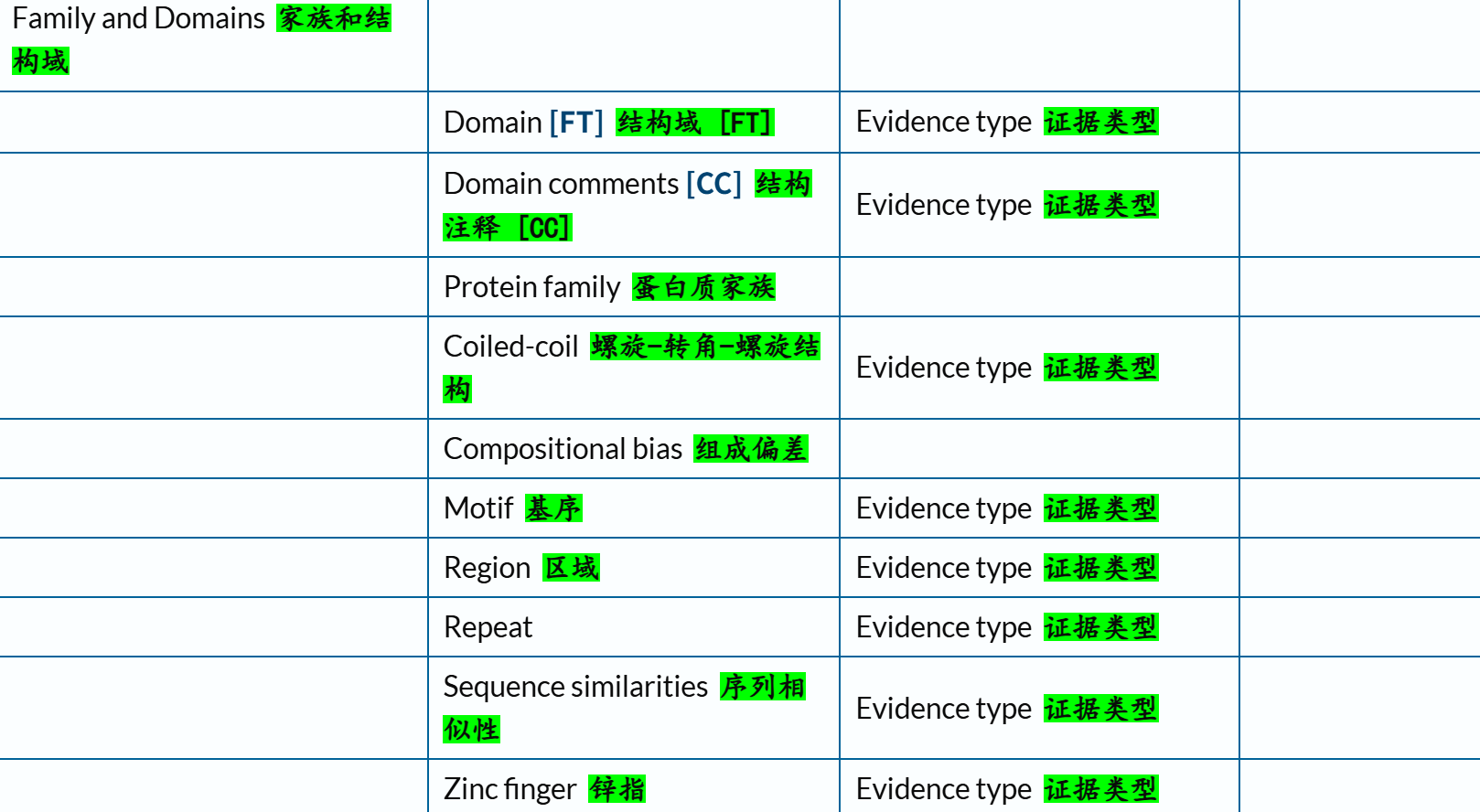
结构域(domain),其实是功能、序列、结构分析的最小单元,也是核心,也是我们这篇博客的核心。
2️⃣如何构建高级搜索?

参考链接来自:
https://www.uniprot.org/help/advanced_search
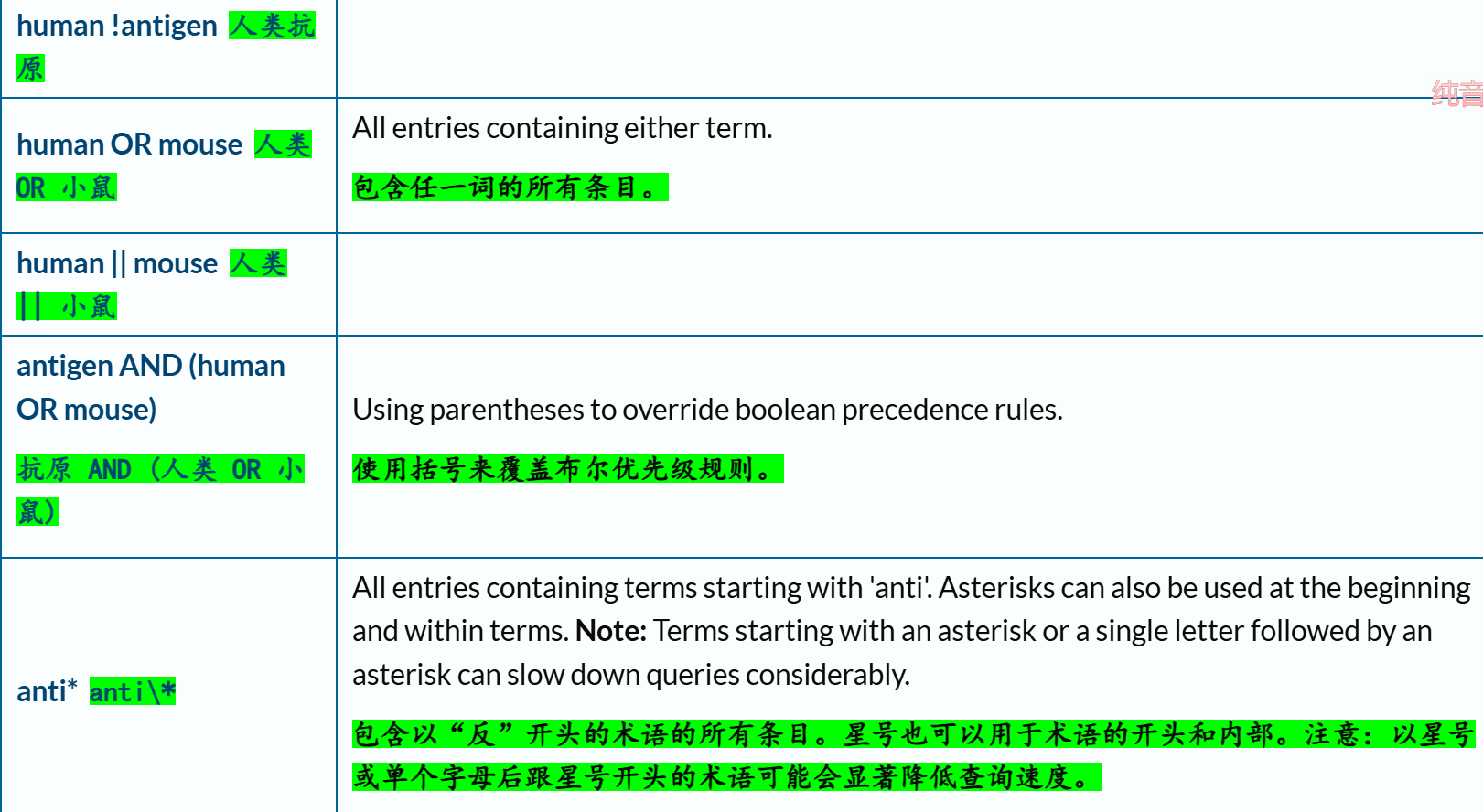
(1)文本搜索:核心在于查询语法(query syntax)
https://www.uniprot.org/help/text-search


上面讲的只是查询语法,语法需要作用对象,就是查询字段(对于query的field有syntax),
关于查询对象,官方文档链接参考(这部分内容很重要,在很多Uniprot数据挖掘中都用得到,搜索任意特定数据的支持查询字段!):
https://www.uniprot.org/help/query-fields
(2)文本搜索的对象:核心在于查询字段(query field)
链接参考:https://www.uniprot.org/help/query-fields

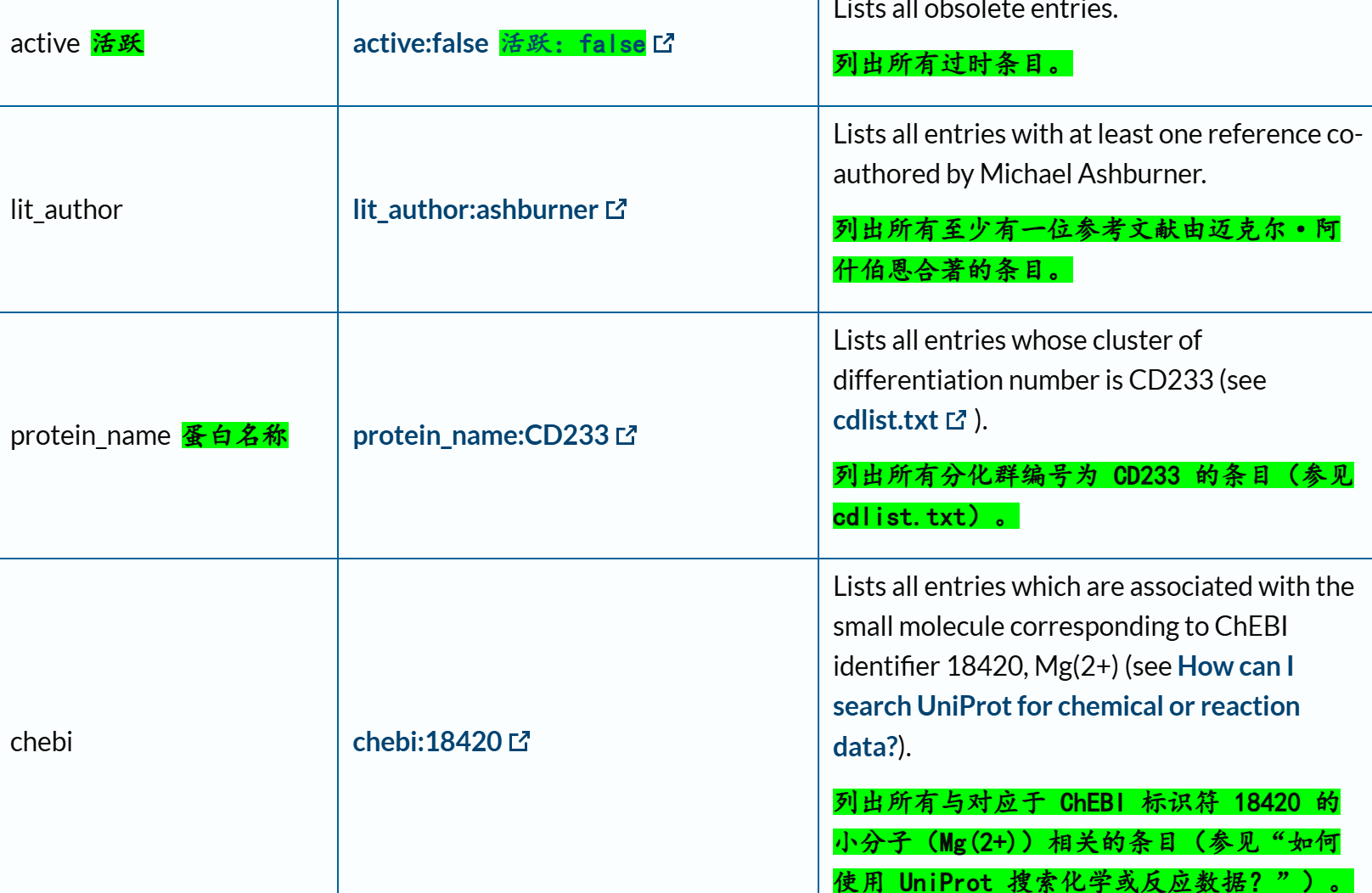
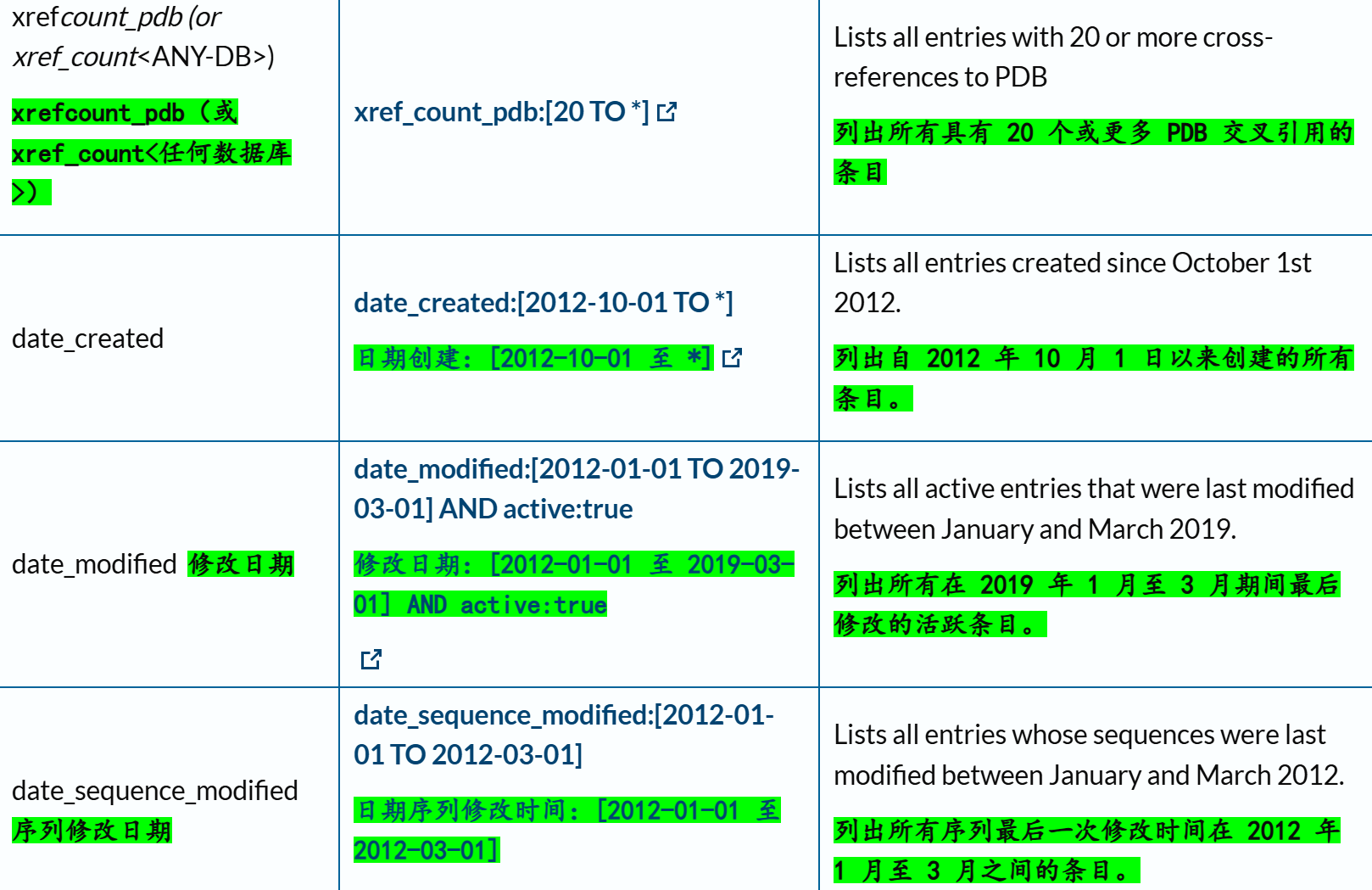
下面这里有一个字段field我经常使用,就是xref,交叉使用其他数据库,因为我们知道Uniprot其实也是集成了很多其他数据库的注释数据,所以有些时候我们需要使用其他数据库的规则或者是内容来限制在Uniprot中的搜索,比如说我想要限制搜索符合Pfam数据库中Krab结构域特征谱的数据,我就需要xref:Pfam的限制语法,然后在Uniprot中搜索。
交叉引用字段的深入,参考:https://www.uniprot.org/help/return_fields_databases

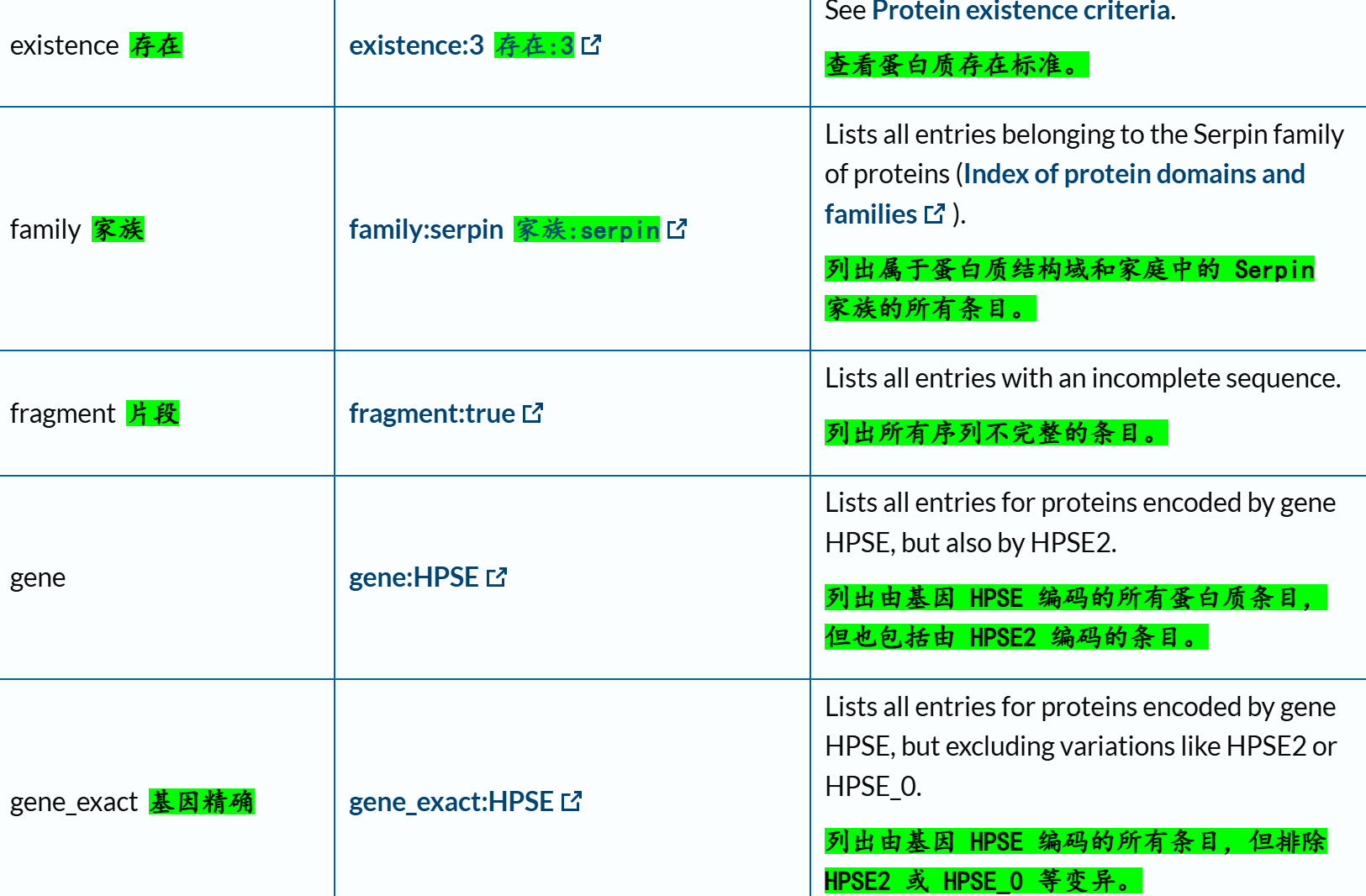
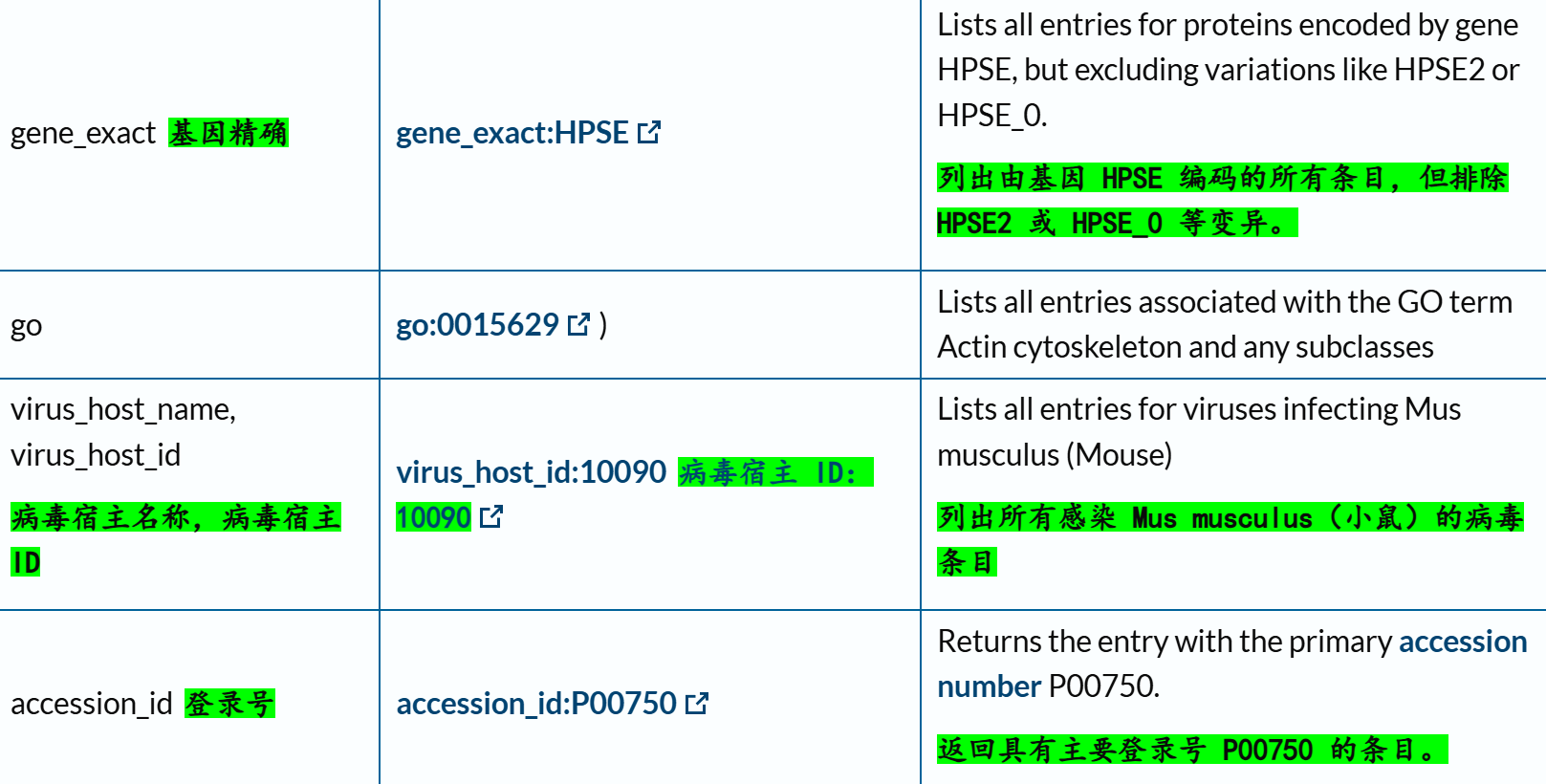



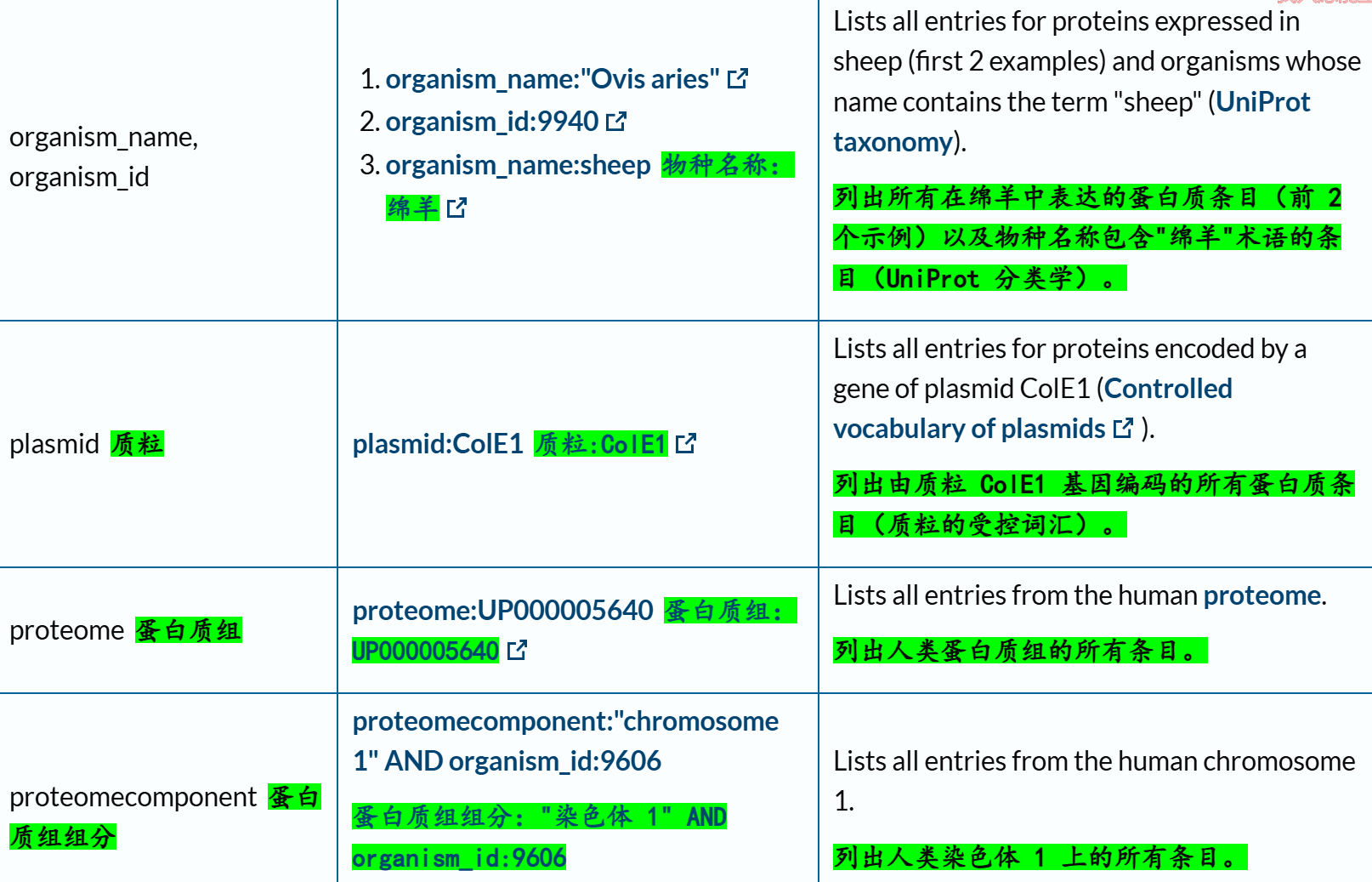
下面的reviewd字段也是非常常用,UniProtKB/Swiss-Prot数据库
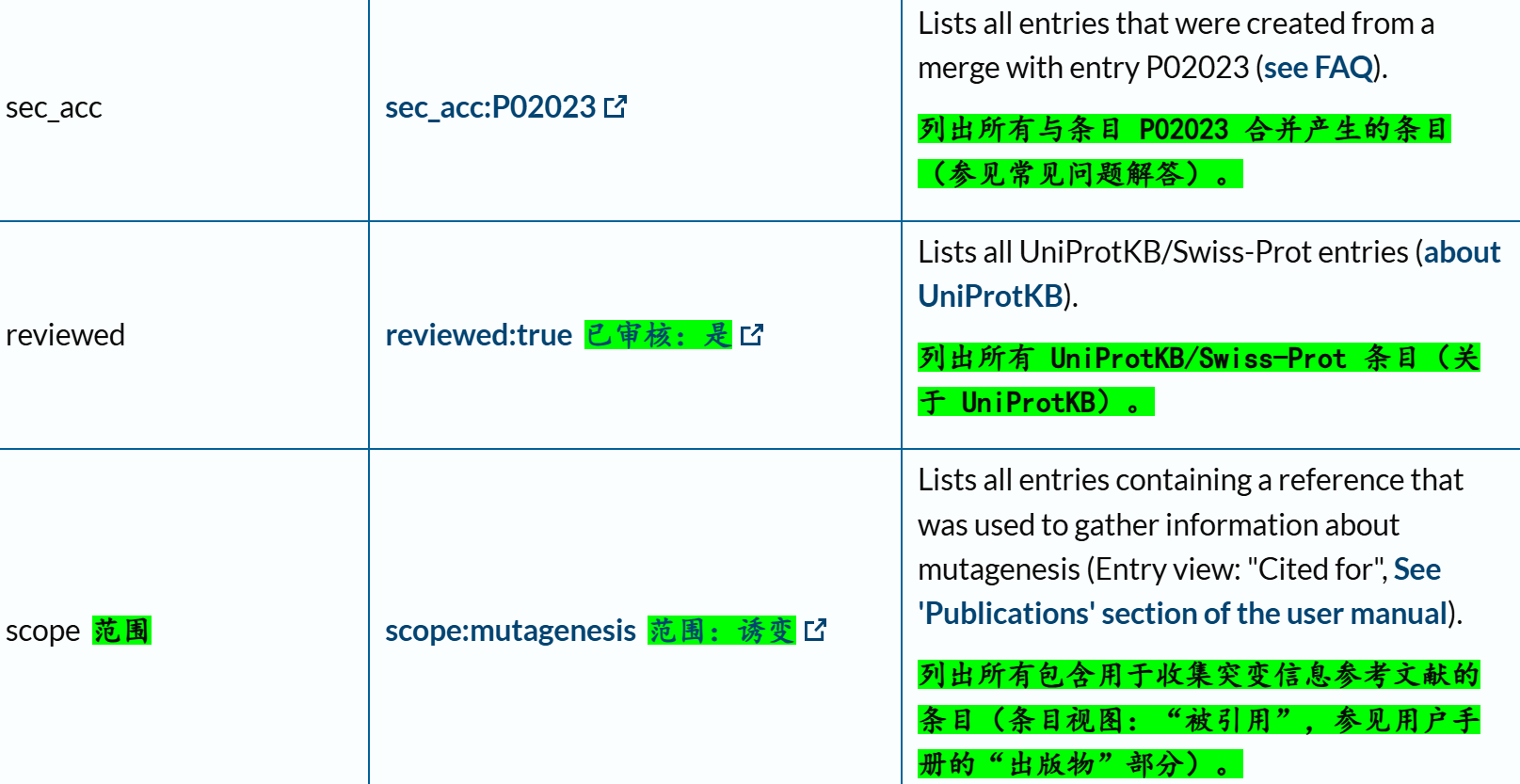

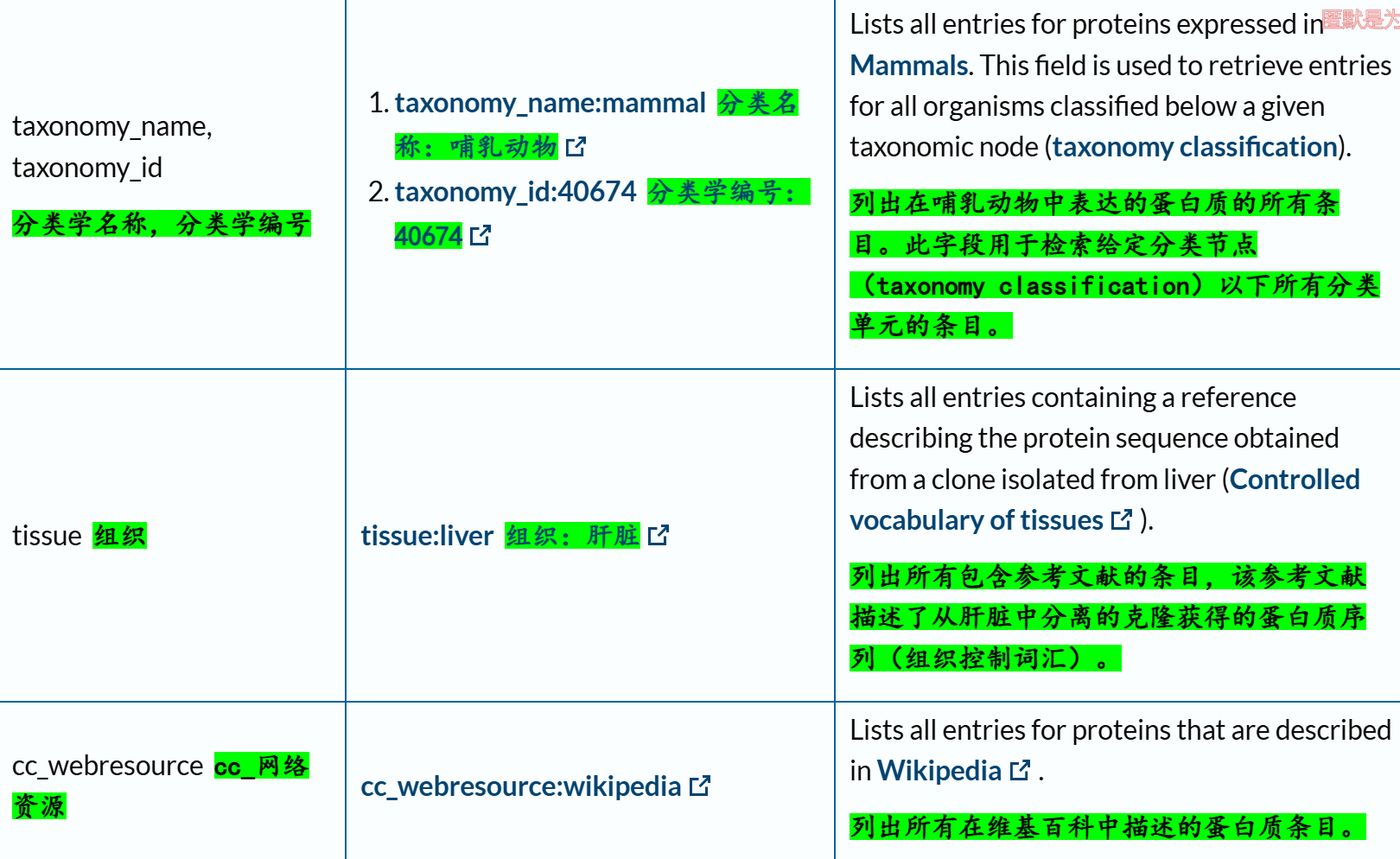
(3)返回字段:return fields
链接参考:https://www.uniprot.org/help/return_fields


curl https://rest.uniprot.org/uniprotkb/search?query=human&fields=accession,xref_proteomes
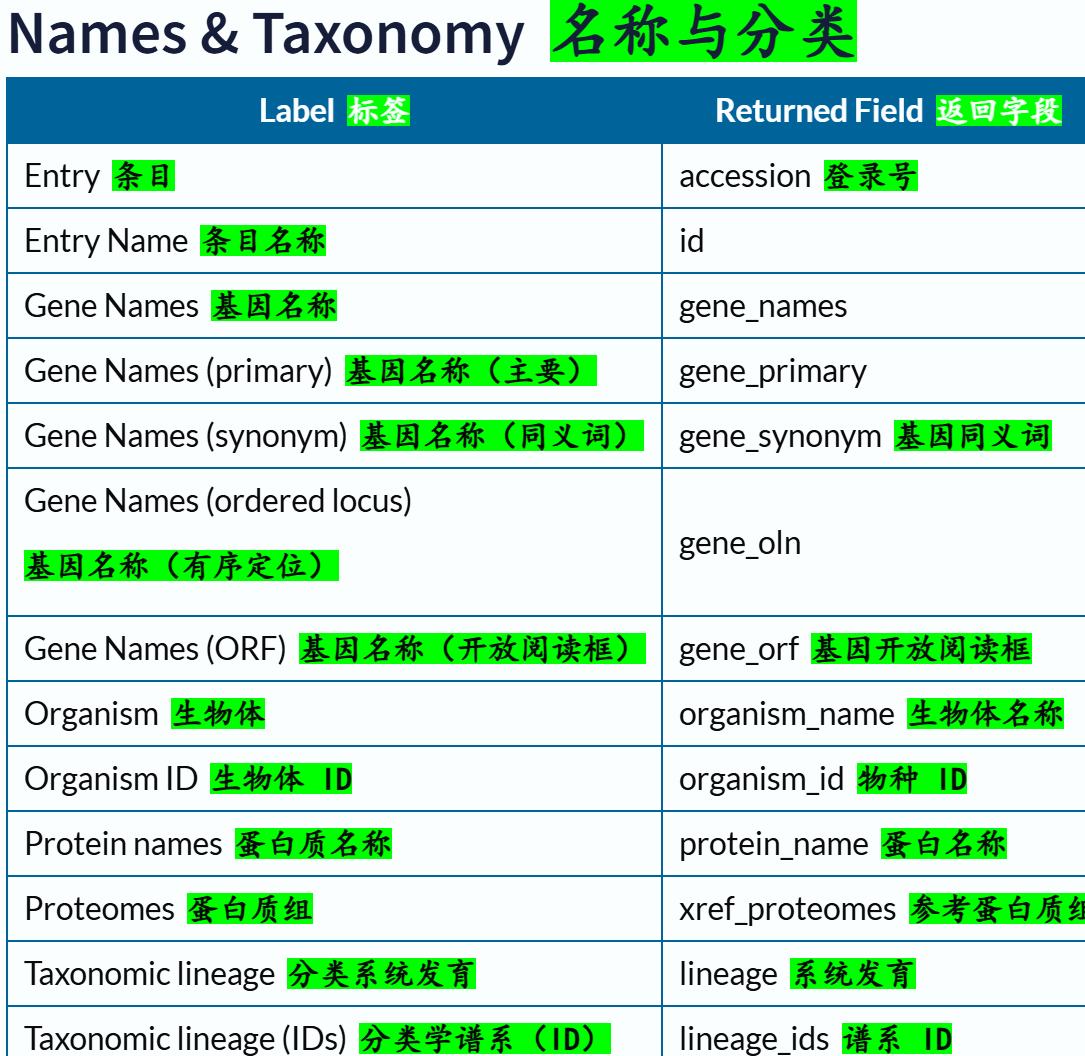


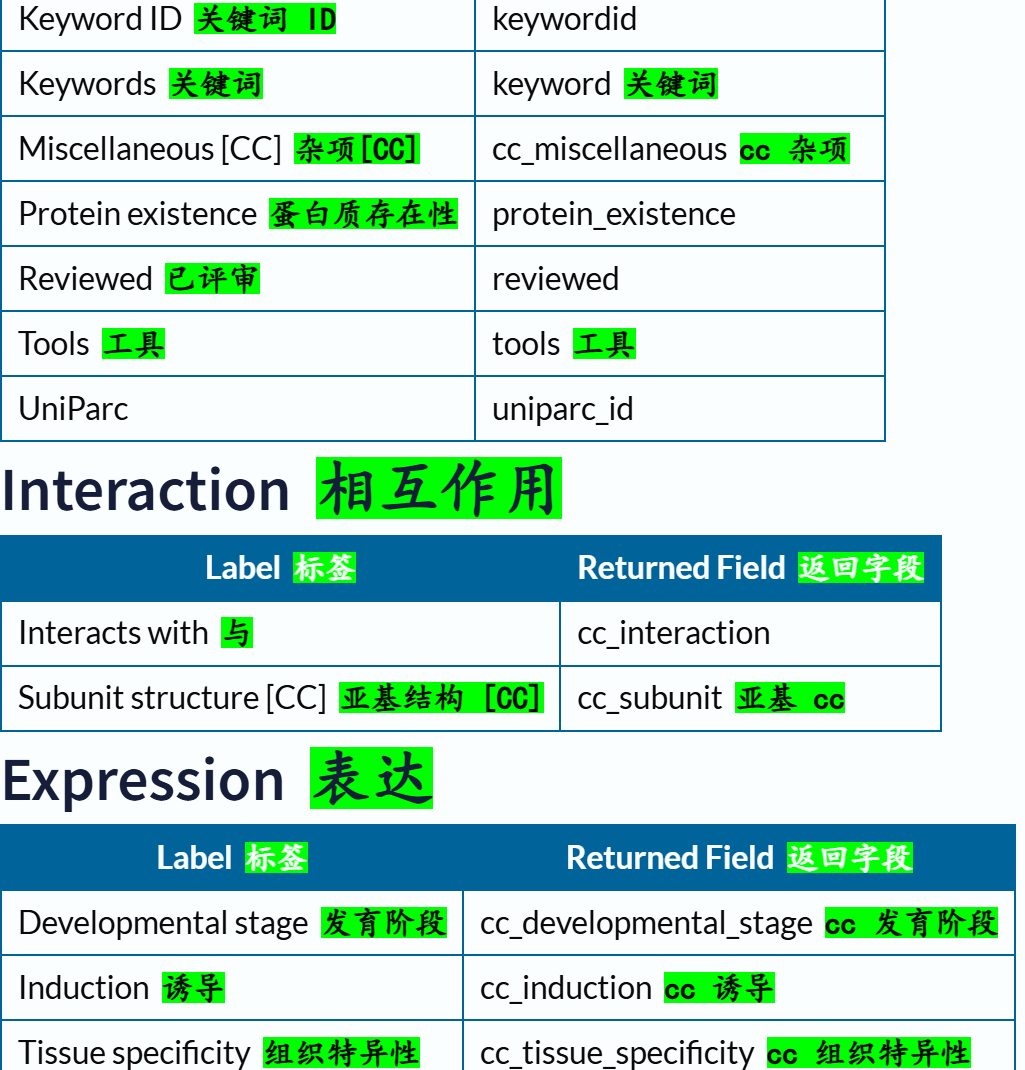
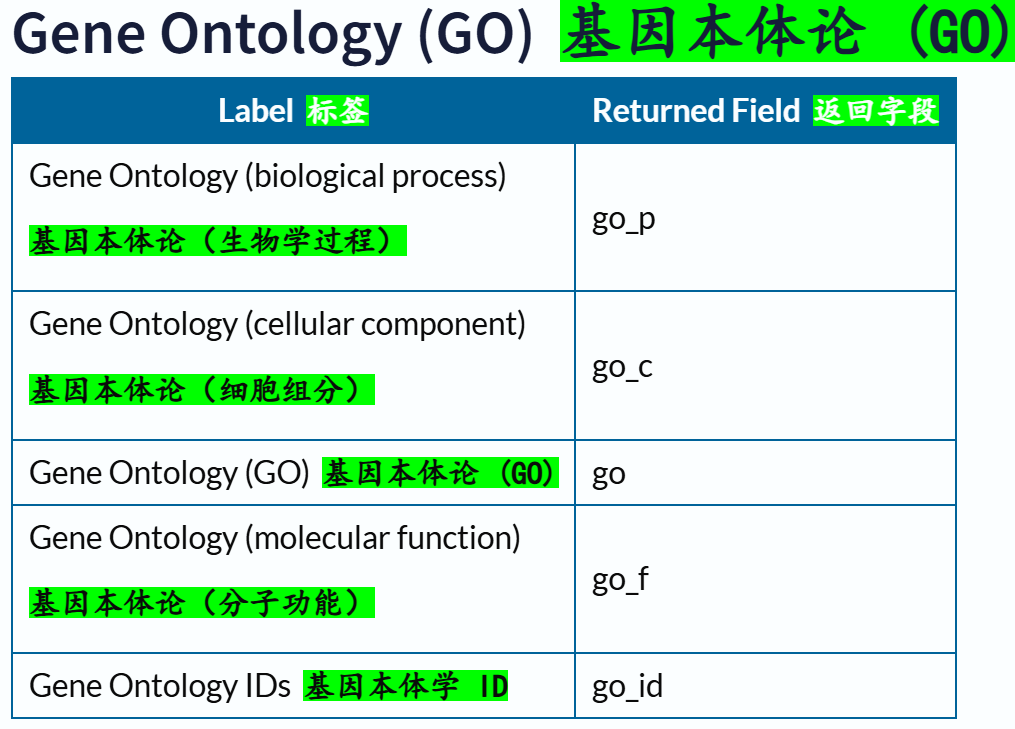
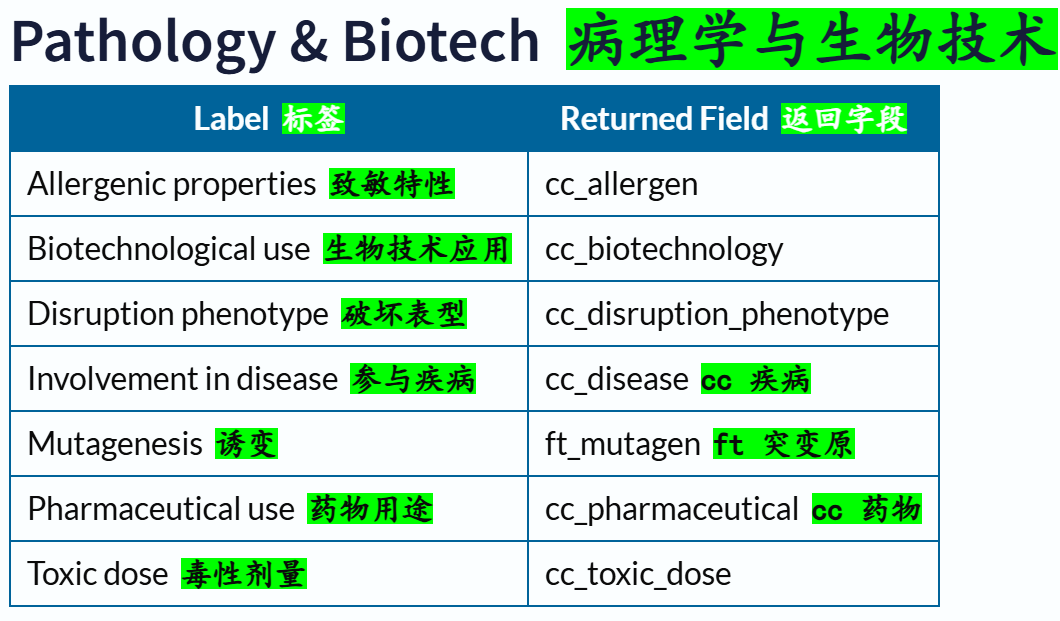

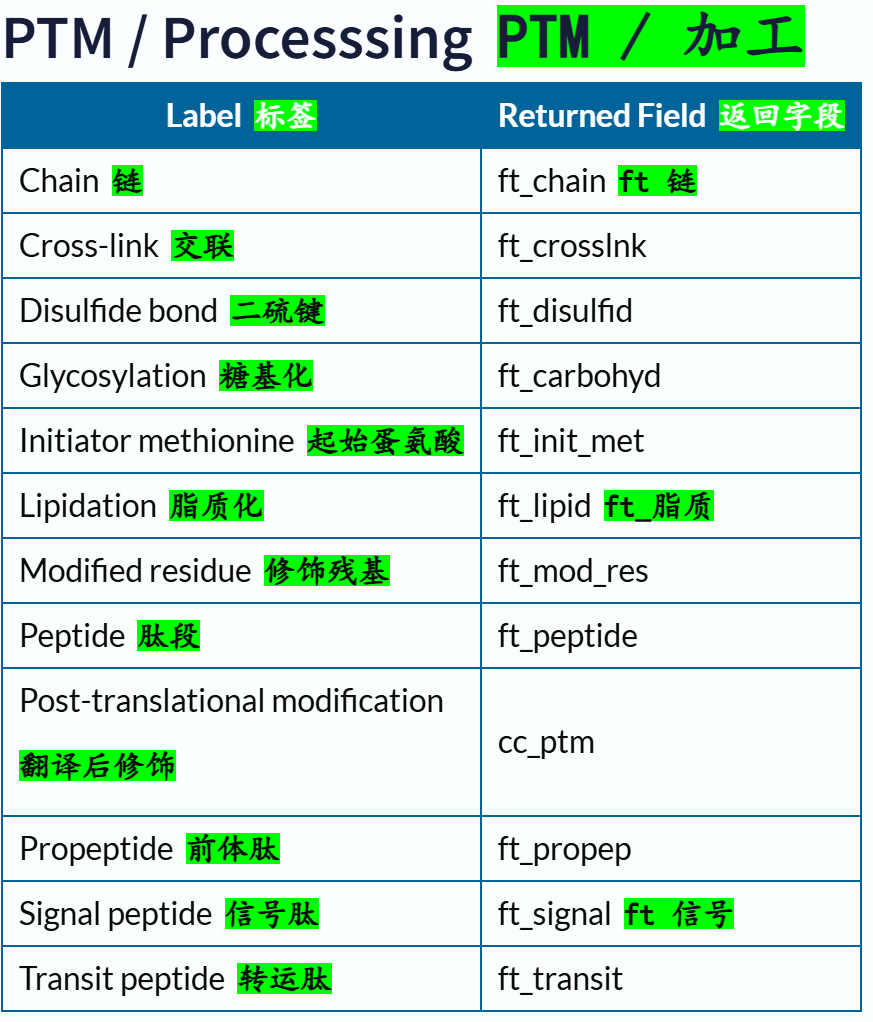



交叉引用部分的返回字段:

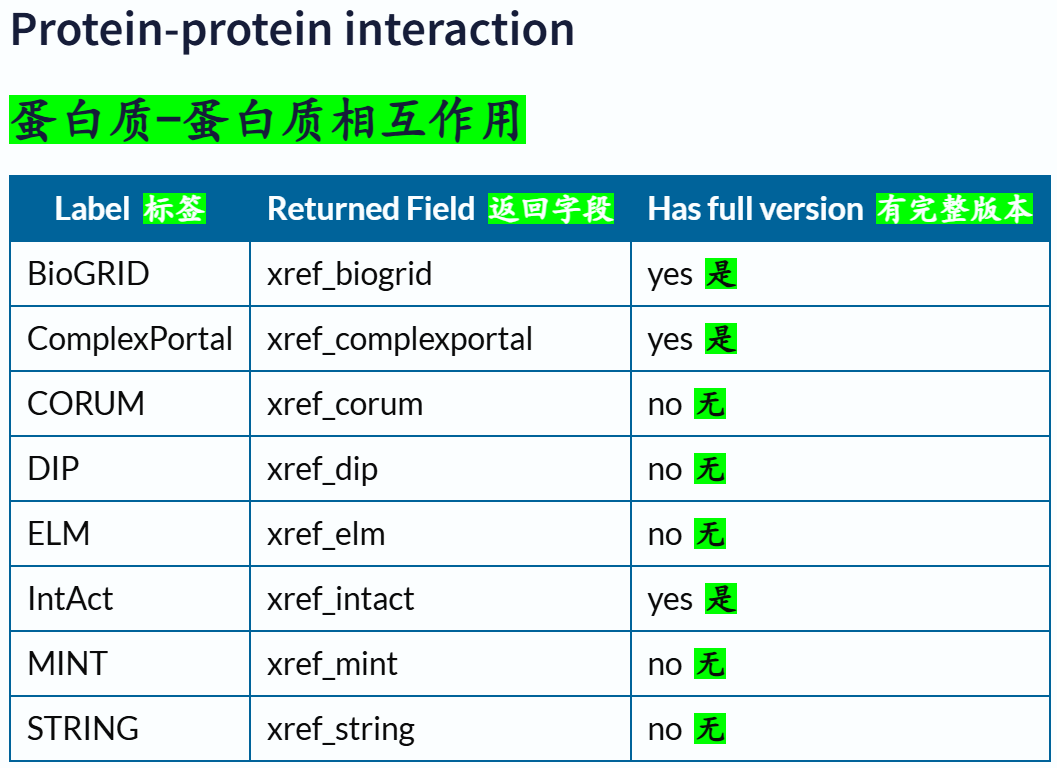
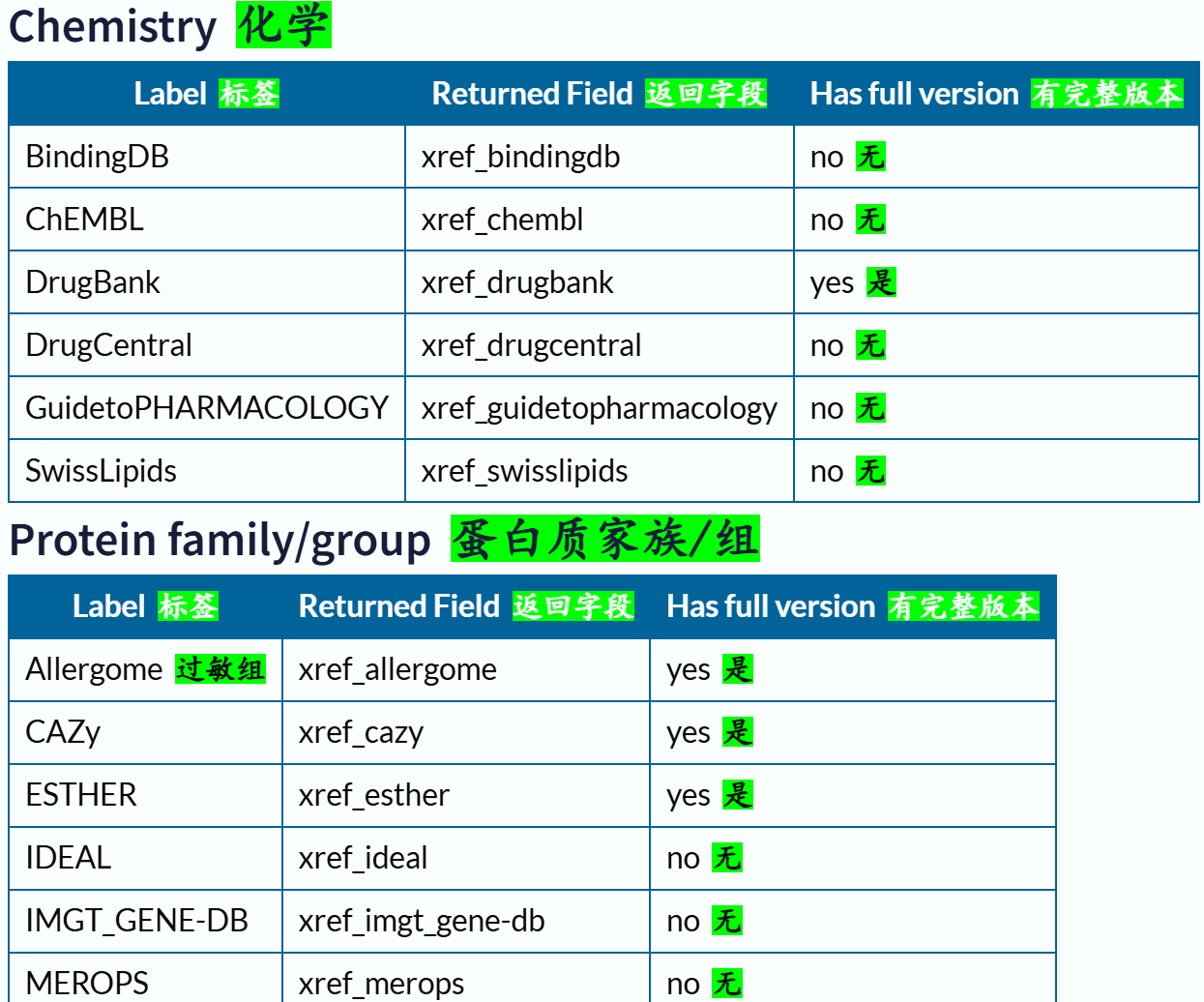
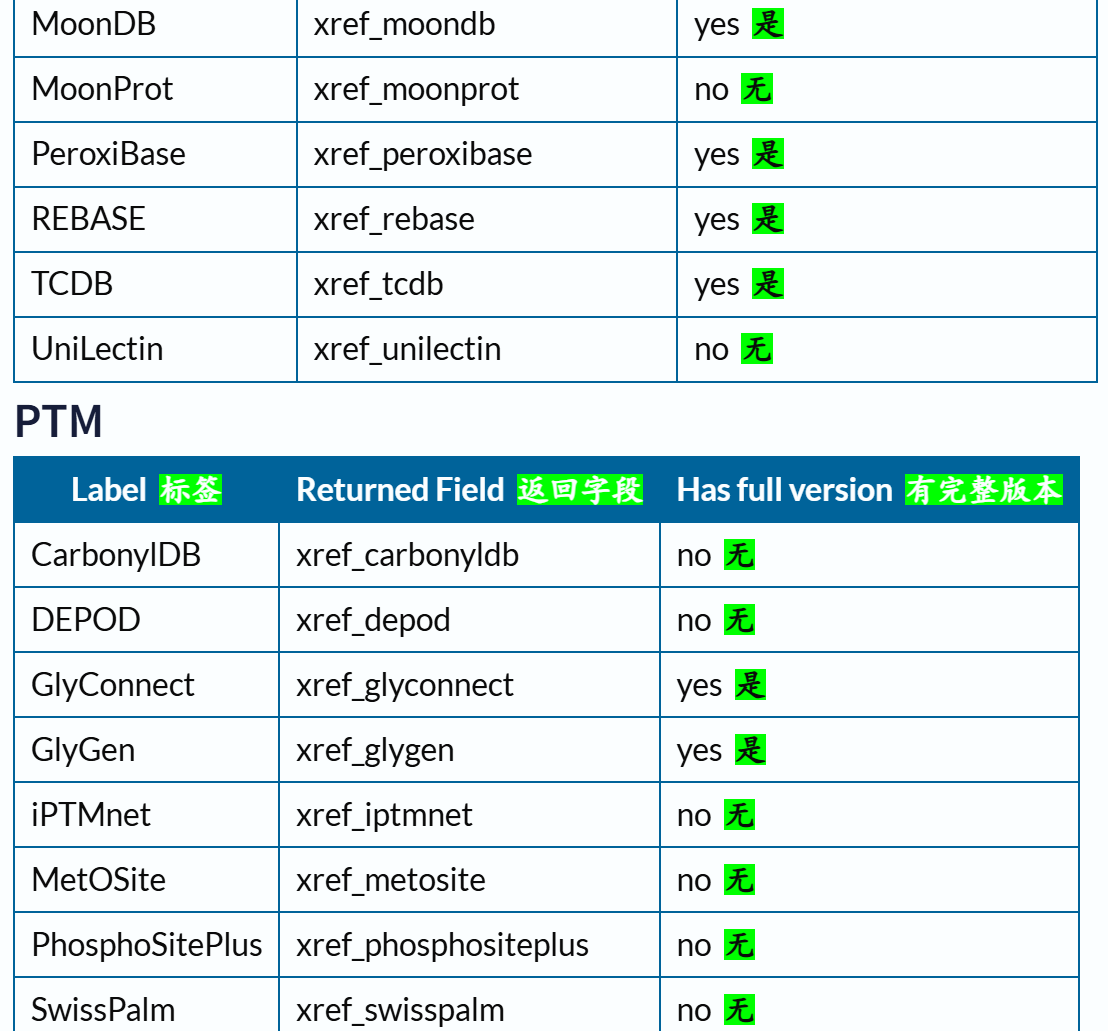
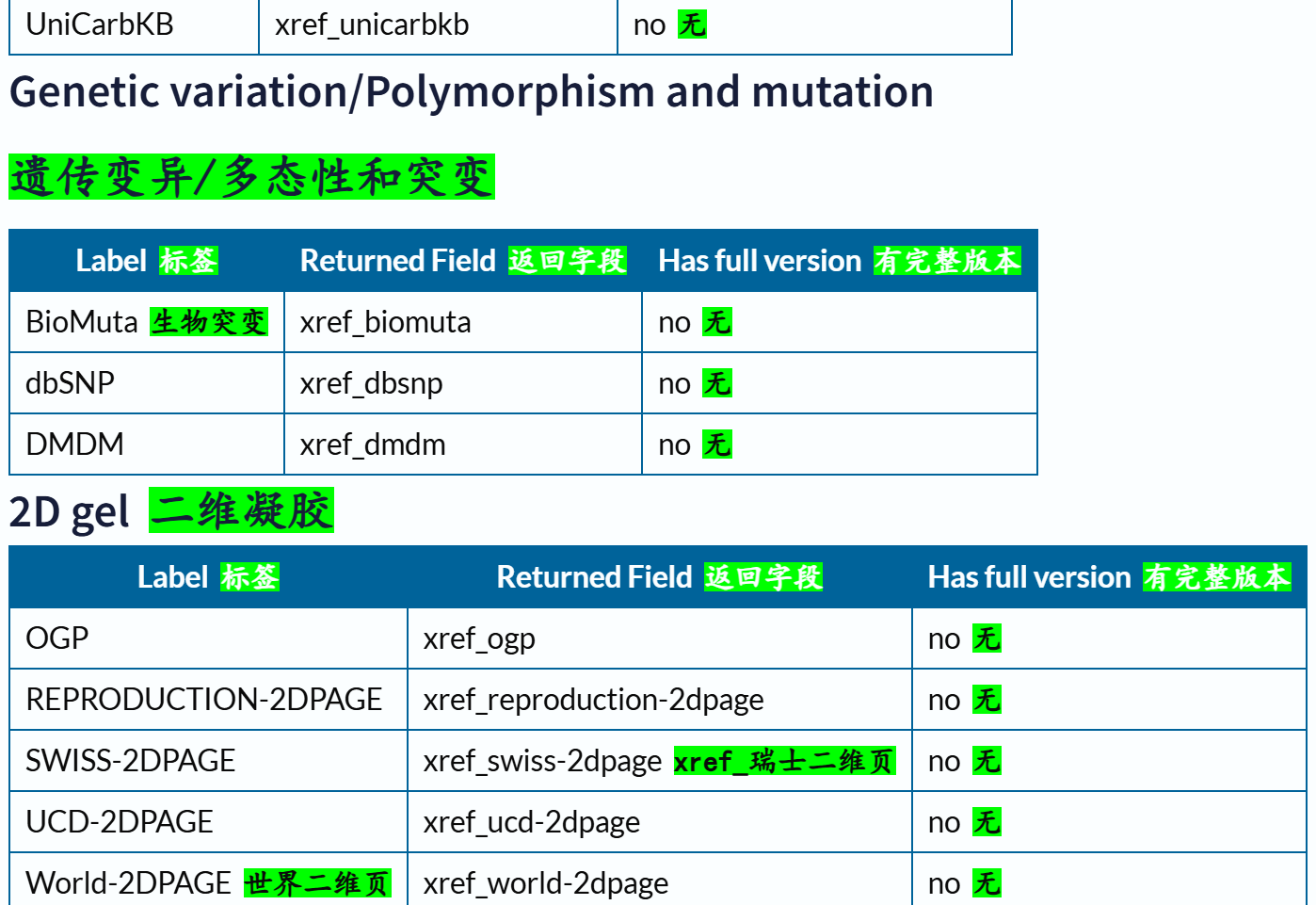
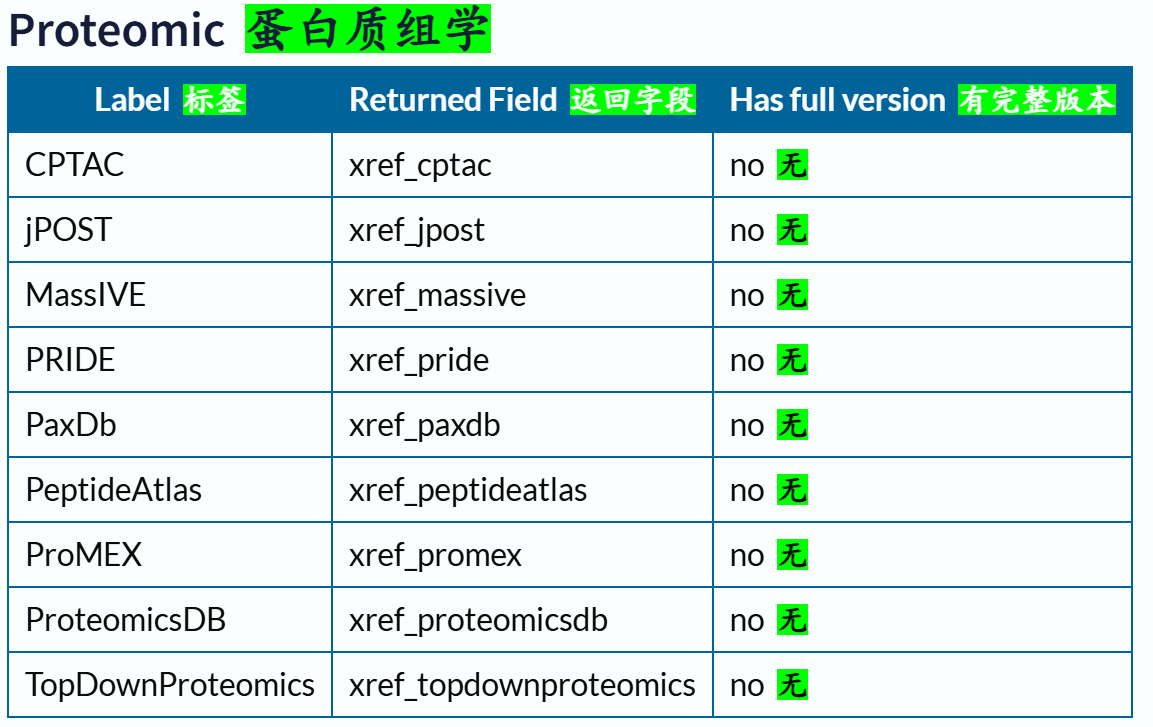
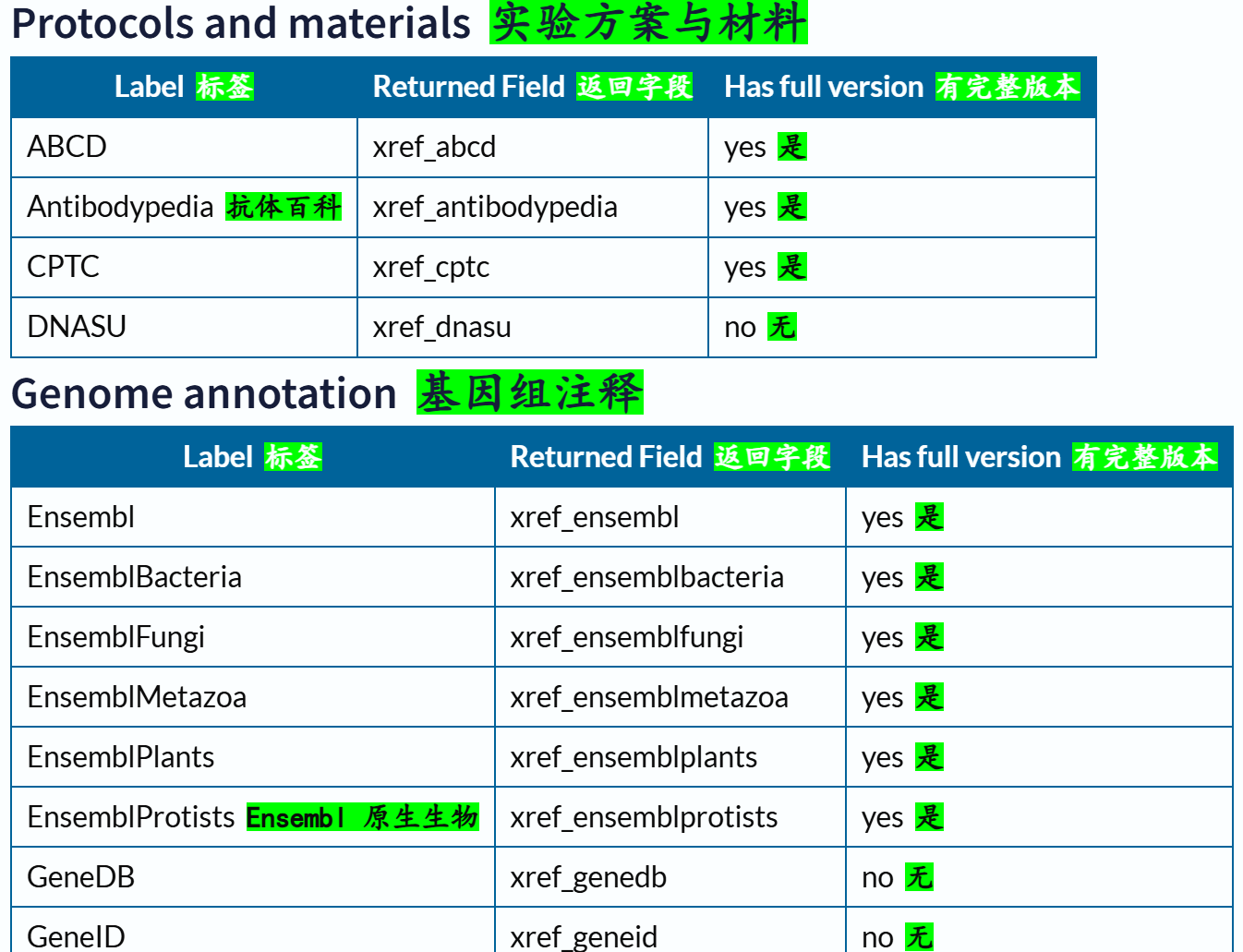

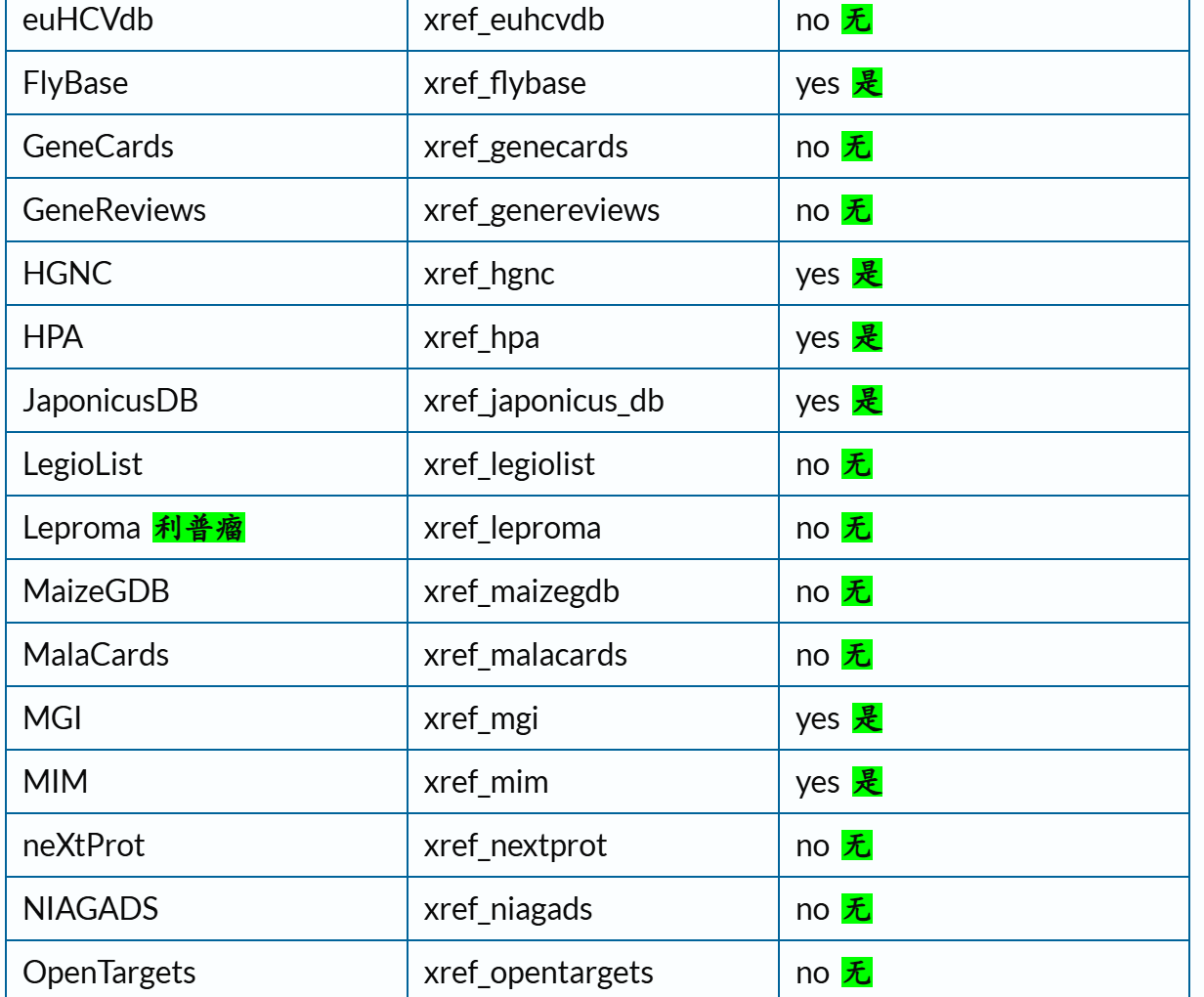
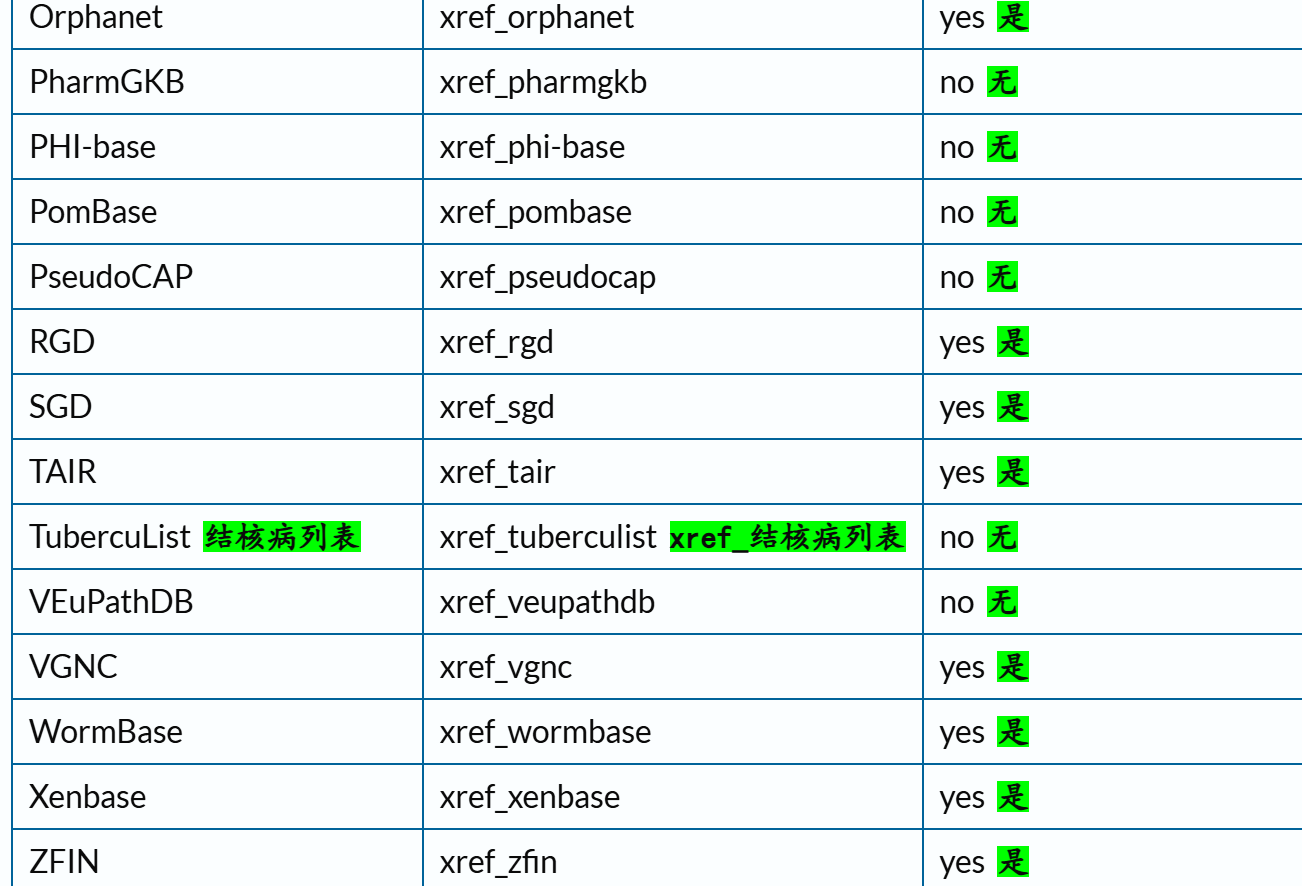
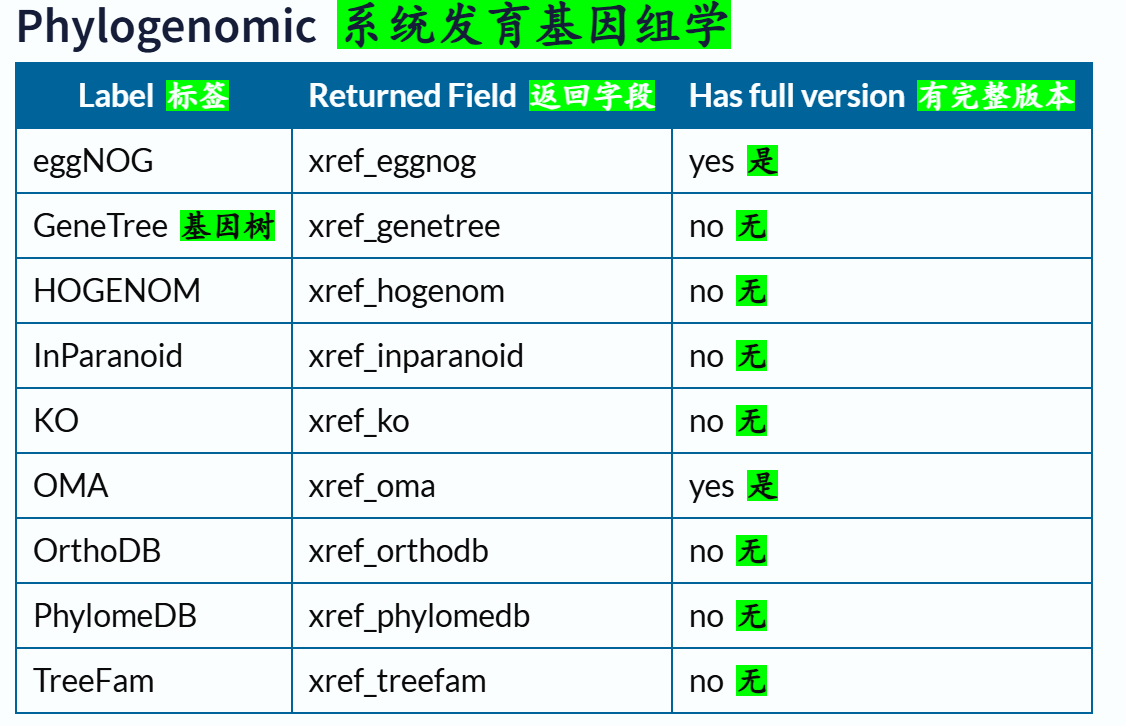
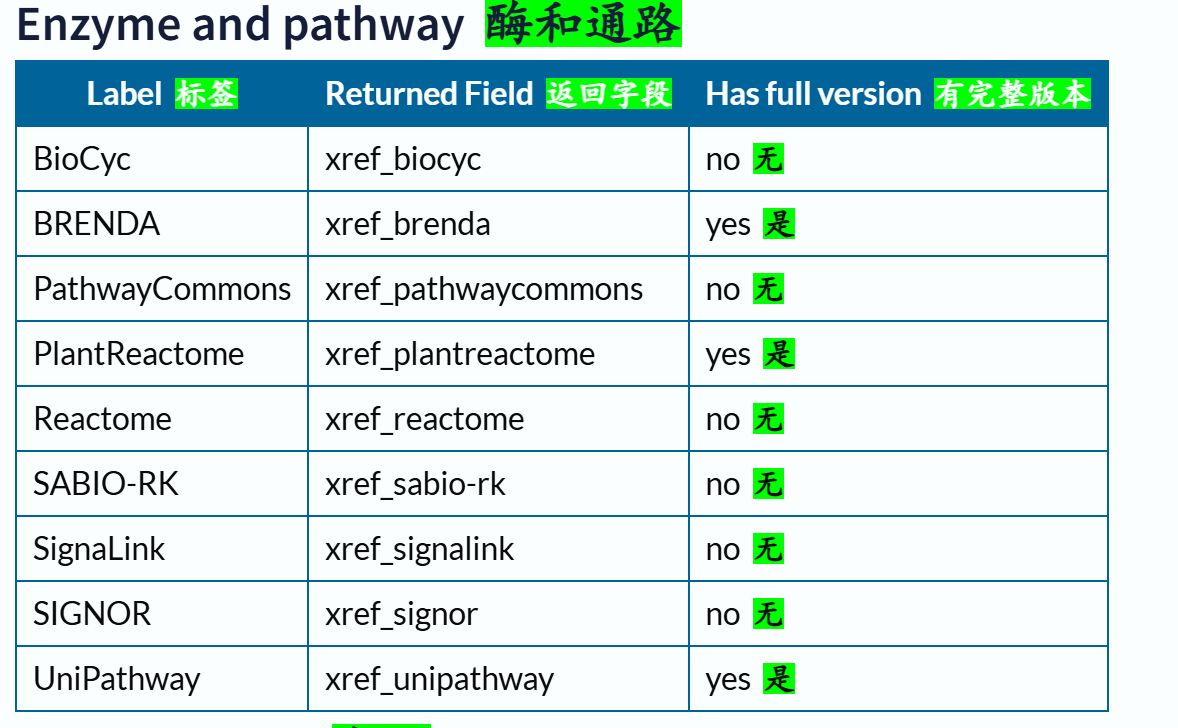
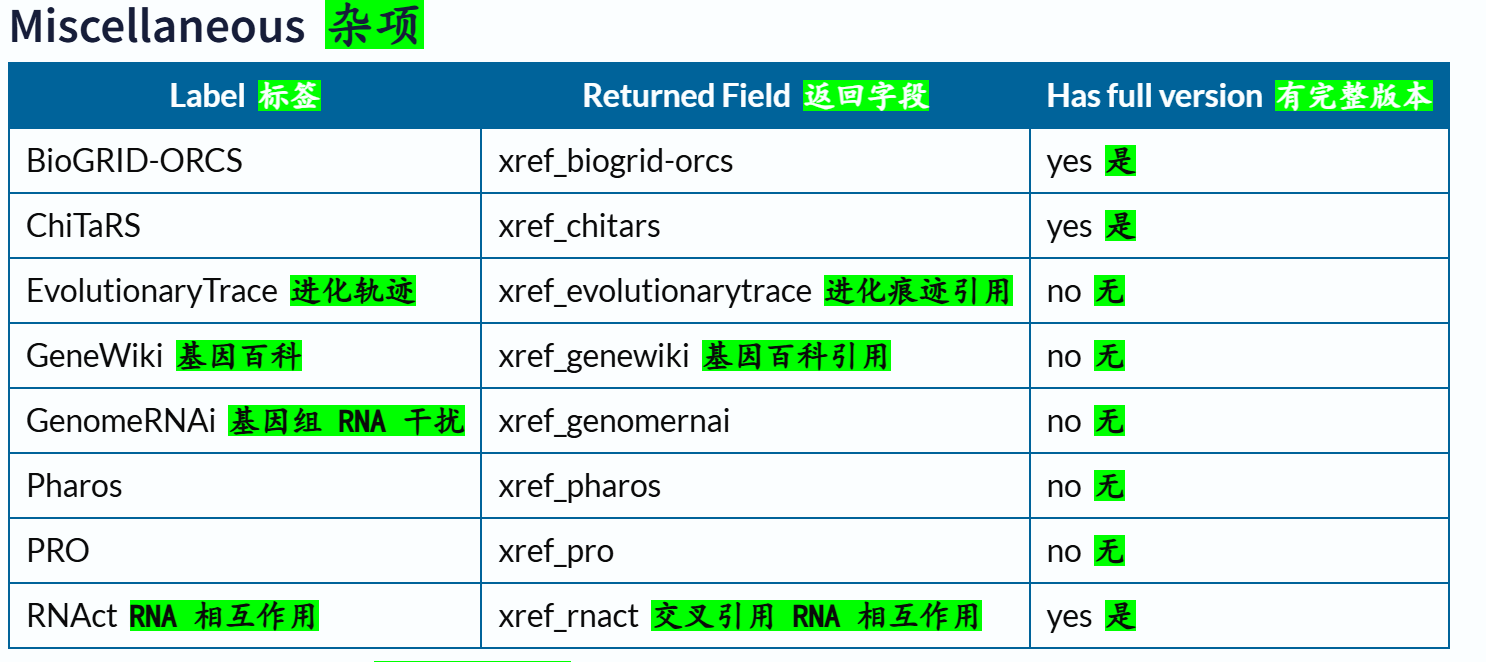
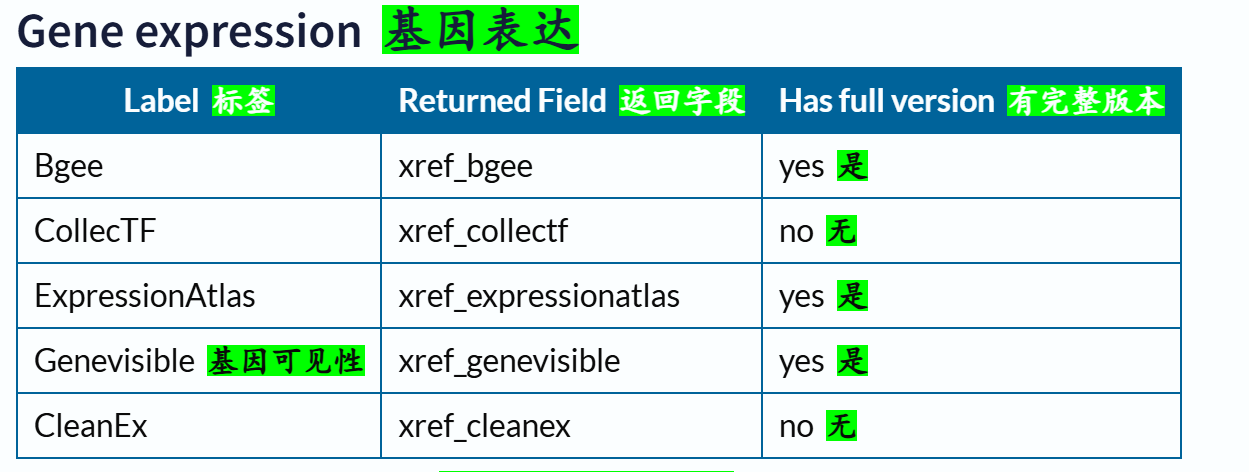

链接参考:https://www.uniprot.org/help/return_fields_databases
查询结果的URL组成,涉及到的查询参数:
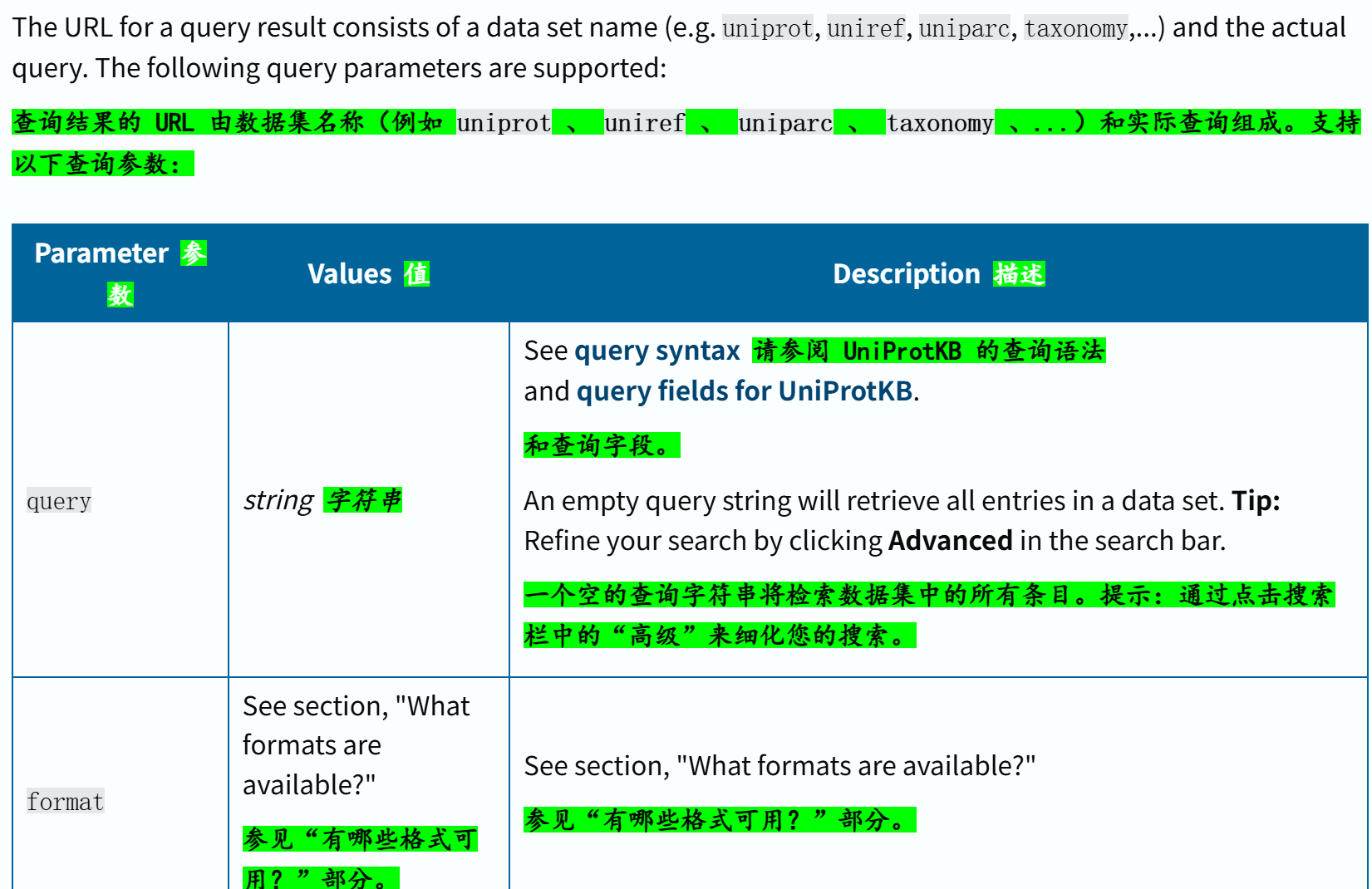
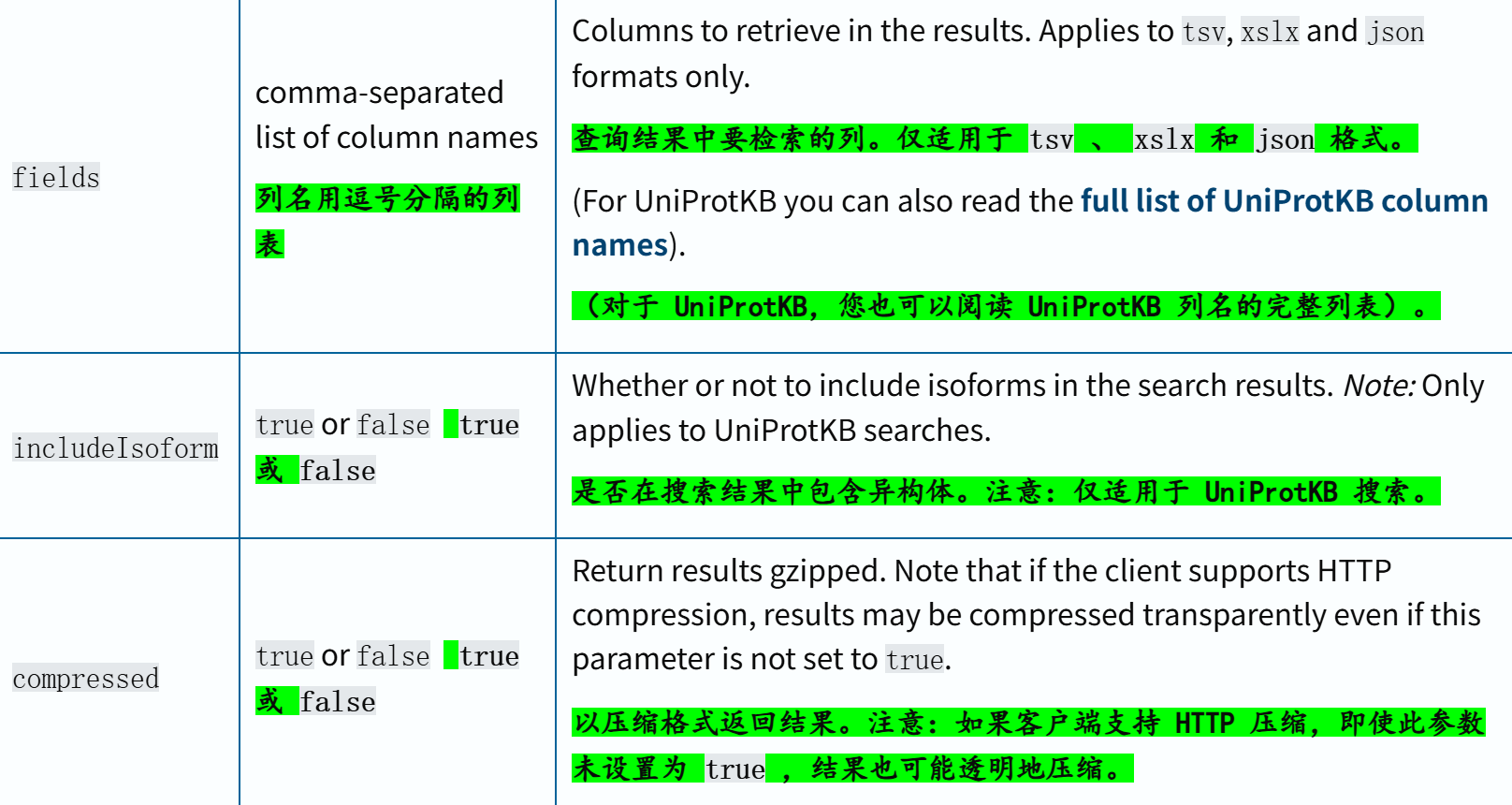

以下示例分别以压缩的 JSON 和制表符分隔值格式检索所有匹配术语’ antigen '的人类条目。
https://rest.uniprot.org/uniprotkb/search?query=organism_id:9606+AND+antigen&format=json&compressed=true
https://rest.uniprot.org/uniprotkb/search?query=organism_id:9606+AND+antigen&format=tsv&compressed=true
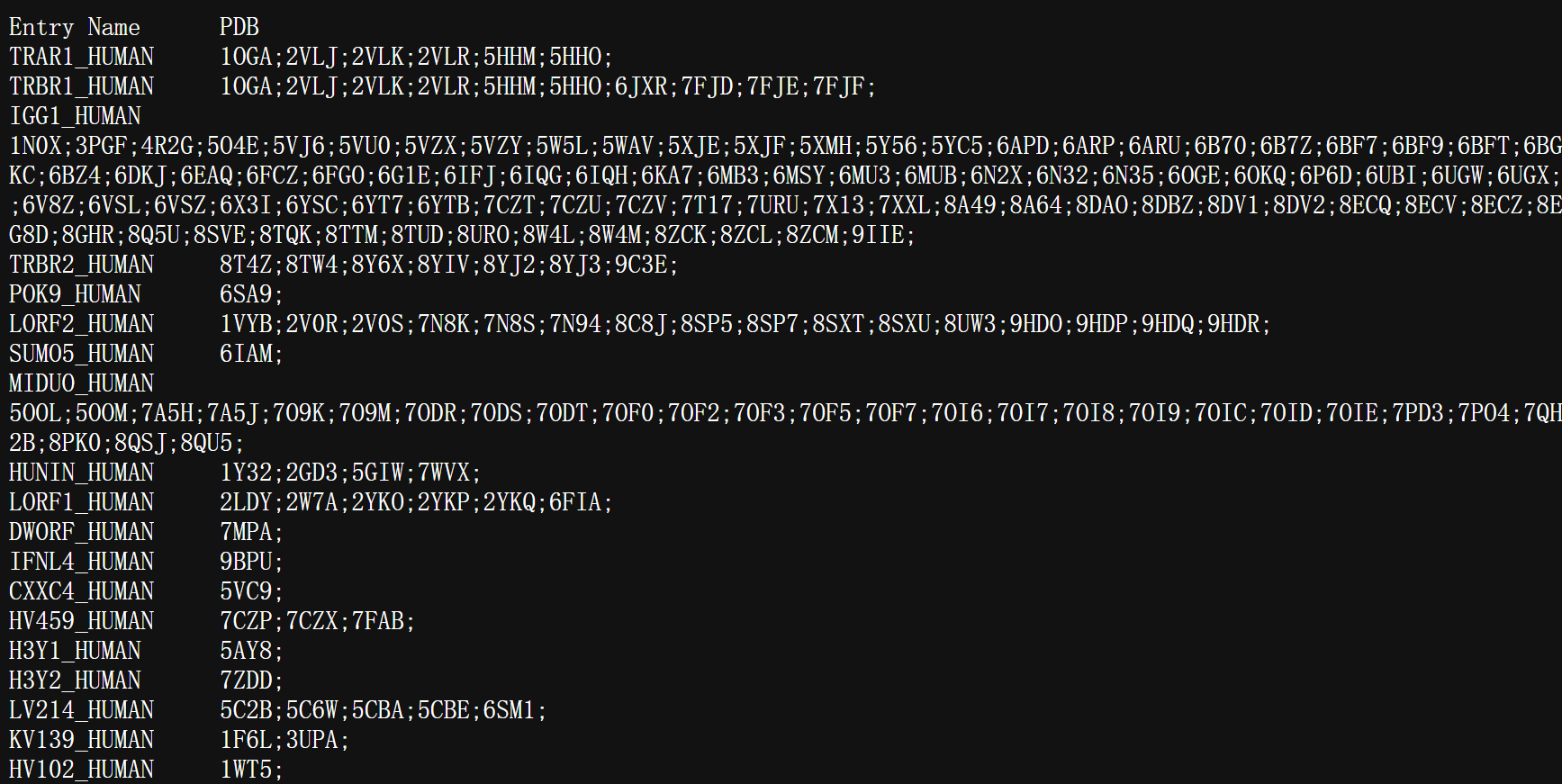
下一个示例以制表符分隔的格式检索所有具有 PDB 交叉引用的人类条目,仅显示 UniProtKB 和 PDB 标识符。
https://rest.uniprot.org/uniprotkb/search?query=organism_id:9606+AND+database:pdb&format=tsv&fields=id,xref_pdb
该链接检索,返回的都是PDB字段
(4)Python示例—在 Python 脚本中立即使用搜索结果
(1)结果数量较少:使用流stream

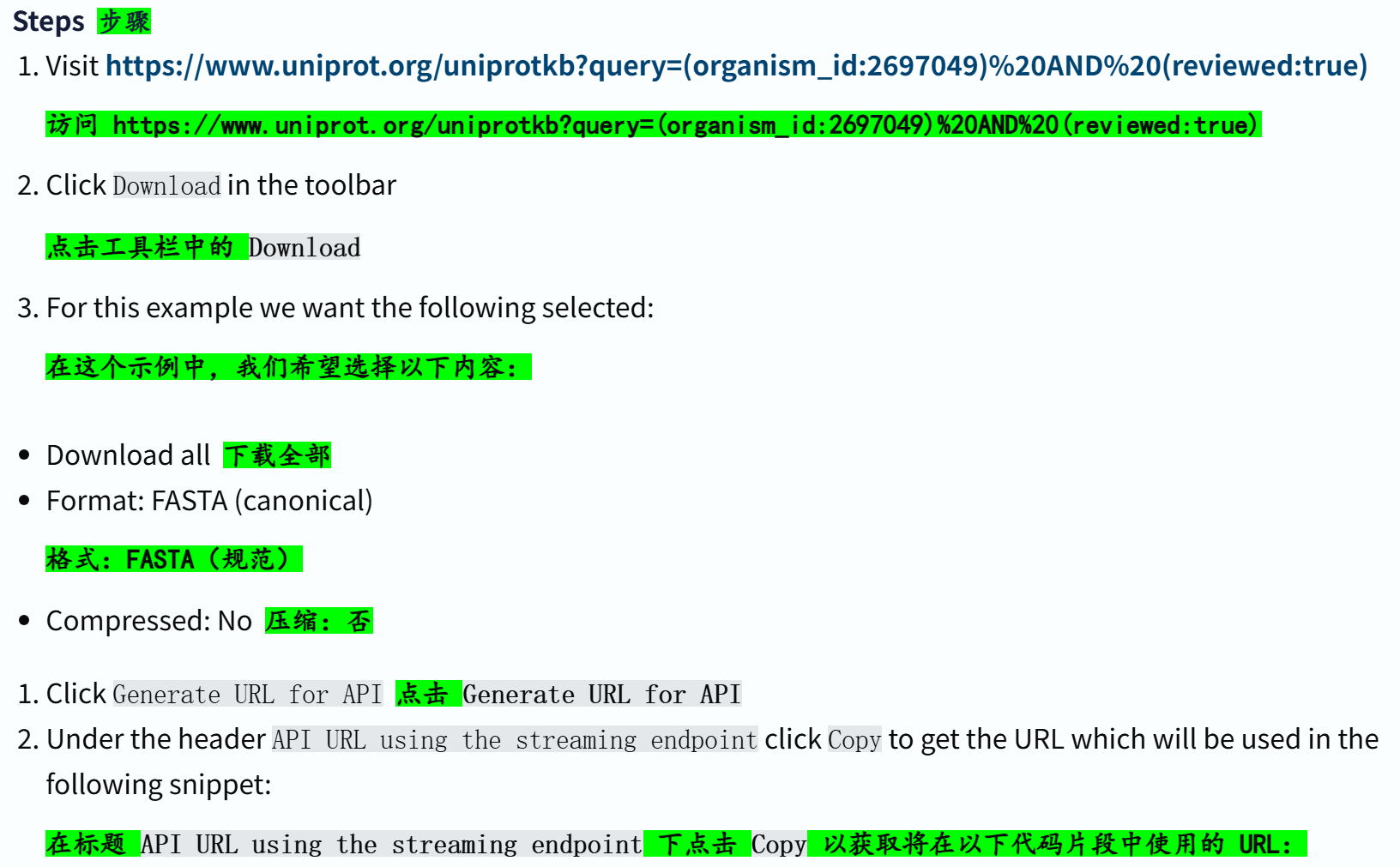
现在我们来实操一下:假设我们想从中获取SARS-COV2病毒蛋白的数据,
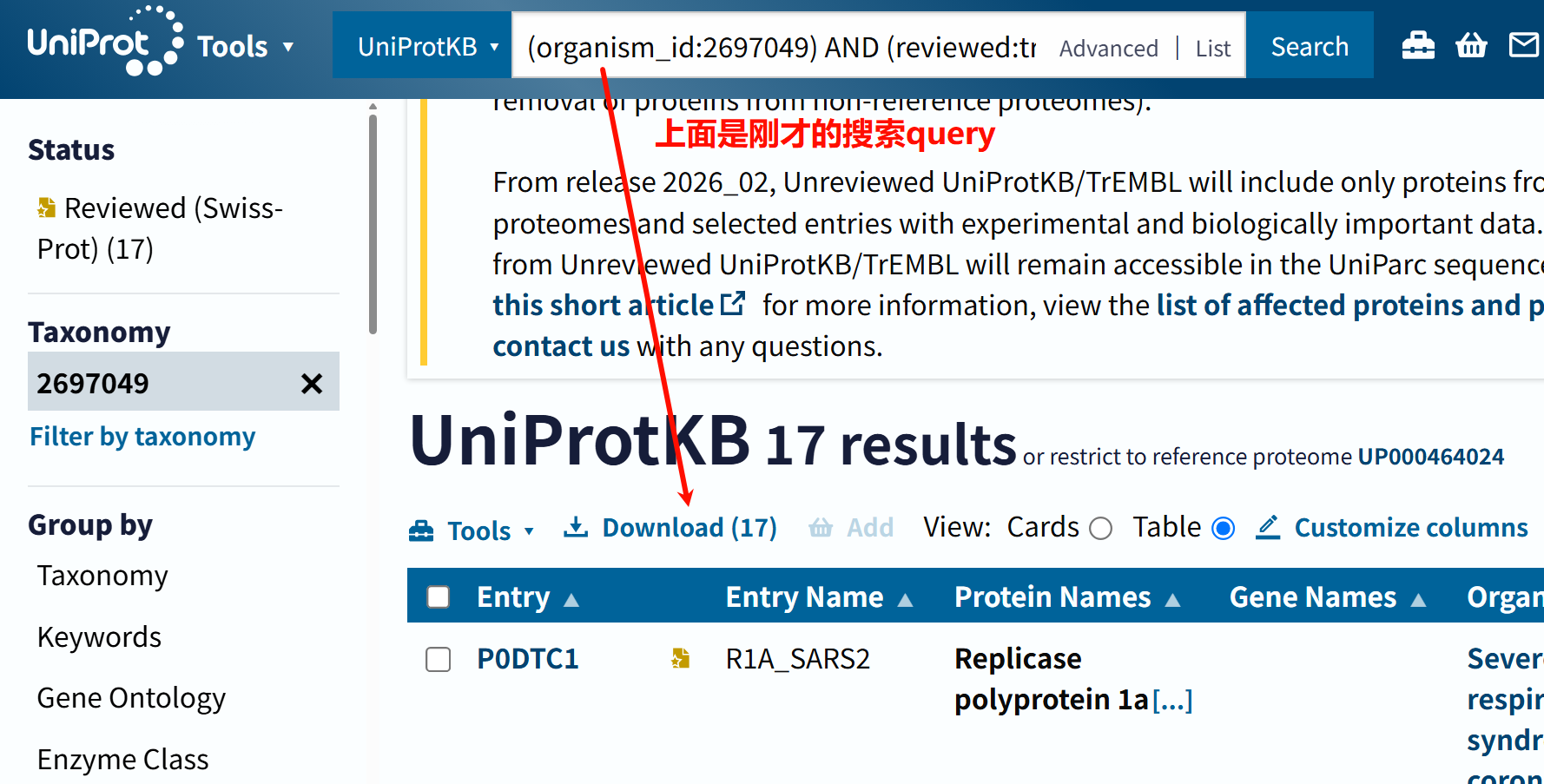
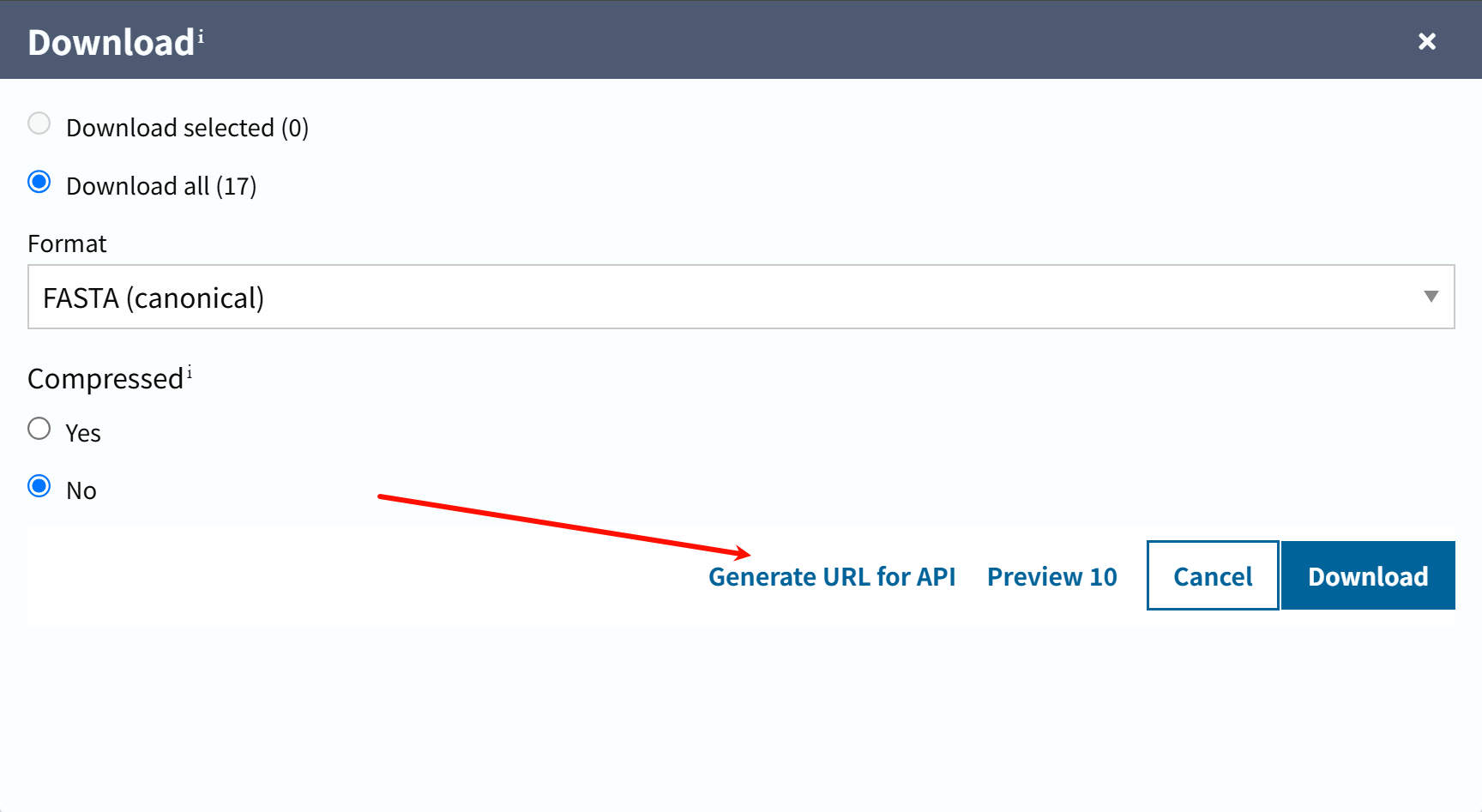
我们可以看到,生成了两个url:
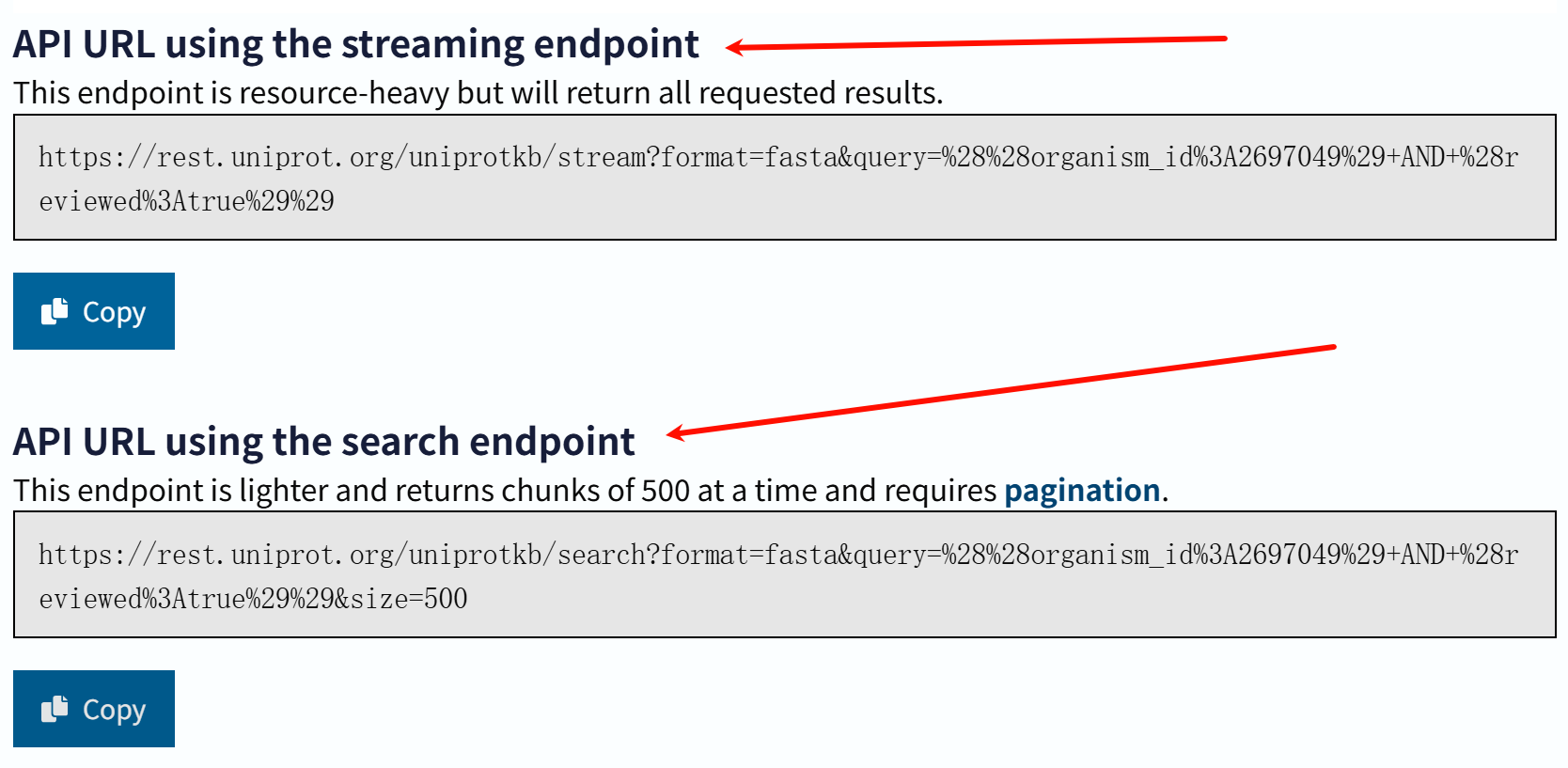
我们只copy第一个,也就是stream流的url,用于下面代码
import requests
url = 'https://rest.uniprot.org/uniprotkb/stream?compressed=false&format=fasta&query=%28organism_id%3A2697049%29%20AND%20%28reviewed%3Atrue%29'
all_fastas = requests.get(url).text
其实代码部分很简单,就是用requests库:
<font style="color:rgba(0, 0, 0, 0.8);">requests</font> 是 Python 中最常用的HTTP 客户端库,用于模拟浏览器 / 客户端与服务器的 HTTP 通信,实现 “发送请求 - 接收响应” 的完整流程。
requests 库的本质是 “简化 HTTP 通信”—— 无需手动处理 TCP 连接、HTTP 协议格式(如请求头、参数拼接)等底层细节,只需调用封装好的函数,即可轻松与 Web 服务器交互。
一般用于,
1️⃣爬取Web资源:向目标网站的 URL 发送请求,获取服务器返回的资源(如网页 HTML、图片二进制数据、JSON 数据等),是 Python 爬虫的 “基石工具”。
2️⃣调用 API 接口(后端 / 数据交互):现代 Web 服务(如微信支付、天气查询、地图服务等)均提供 API 接口(本质是特定 URL),通过发送请求并携带参数,可获取结构化数据(通常是 JSON 格式),用于程序间的数据交互。
3️⃣模拟用户行为(自动化交互):通过设置请求头(如模拟浏览器标识、登录 Cookie),可模拟真实用户的操作。
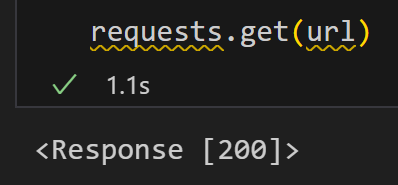
200就是OK

可以看到,返回结果就是一堆fasta格式文件

此时, all_fastas 将包含一个包含所有 FASTA 序列的单个字符串。从这里,我们可以创建一个 FASTA 序列列表,并找到所有标题中提及 SPIKE 的序列:
下面会用到正则表达式的功能,所以需要导入re库
import re
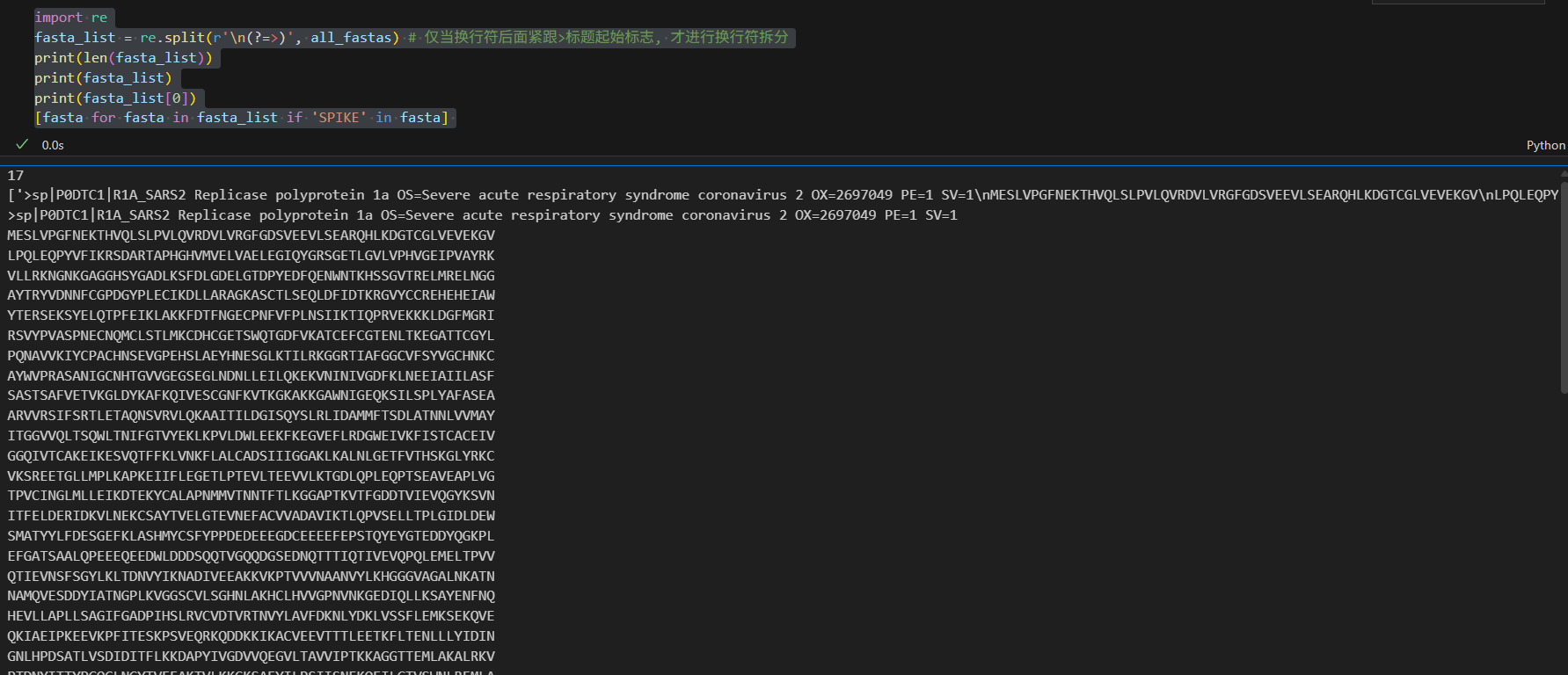
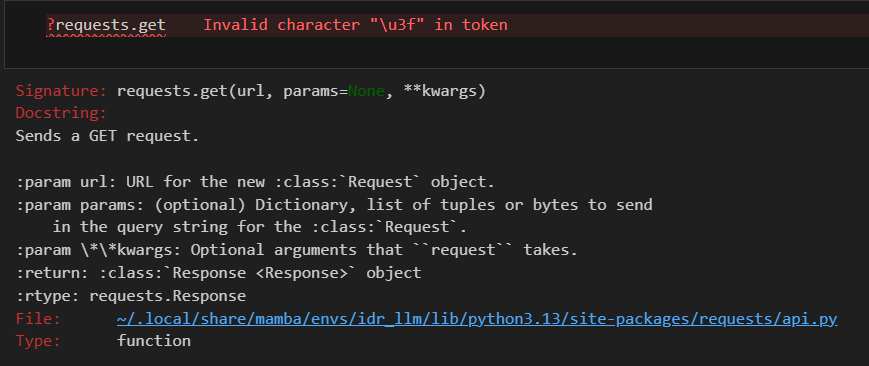
fasta_list = re.split(r'\n(?=>)', all_fastas) # 仅当换行符后面紧跟>标题起始标志, 才进行换行符拆分
print(len(fasta_list))
print(fasta_list)
print(fasta_list[0])
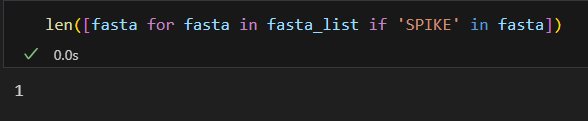
[fasta for fasta in fasta_list if 'SPIKE' in fasta]
效果就是一共17条序列,
拆分开来之后的每一个fasta条目,我们只看其中的包含spike的蛋白:

实际上只有一条:
(2)结果数量较多:使用分页pagination
处理大量结果时,最好使用分页端点(endpoint),

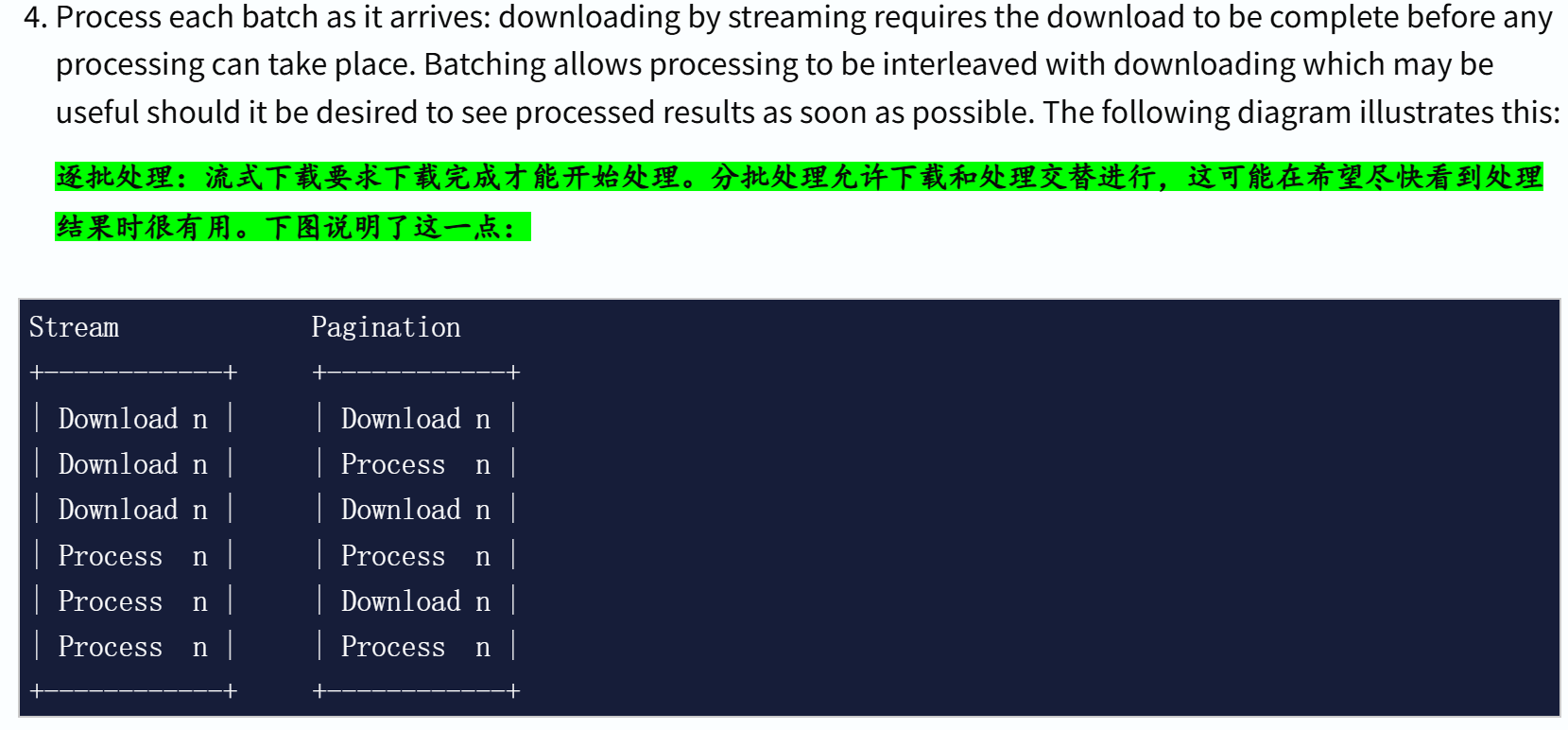

同样的,我们使用示例来展示:通过 UniProt 的 REST API 分页获取 “已审核的胰岛素相关条目” 数据(包含单词 Insulin 的已评审 UniProtKB 条目,并按交互次数最多进行排序)


参考链接:https://www.uniprot.org/uniprotkb?query=Insulin%20AND%20(reviewed:true)
同样的按照前面那样进行实操,在界面上获取url:
这个时候我们选取下面那个url的api,其实我们可以看到,只要尺寸接近500时,就可以考虑使用分页端点了:
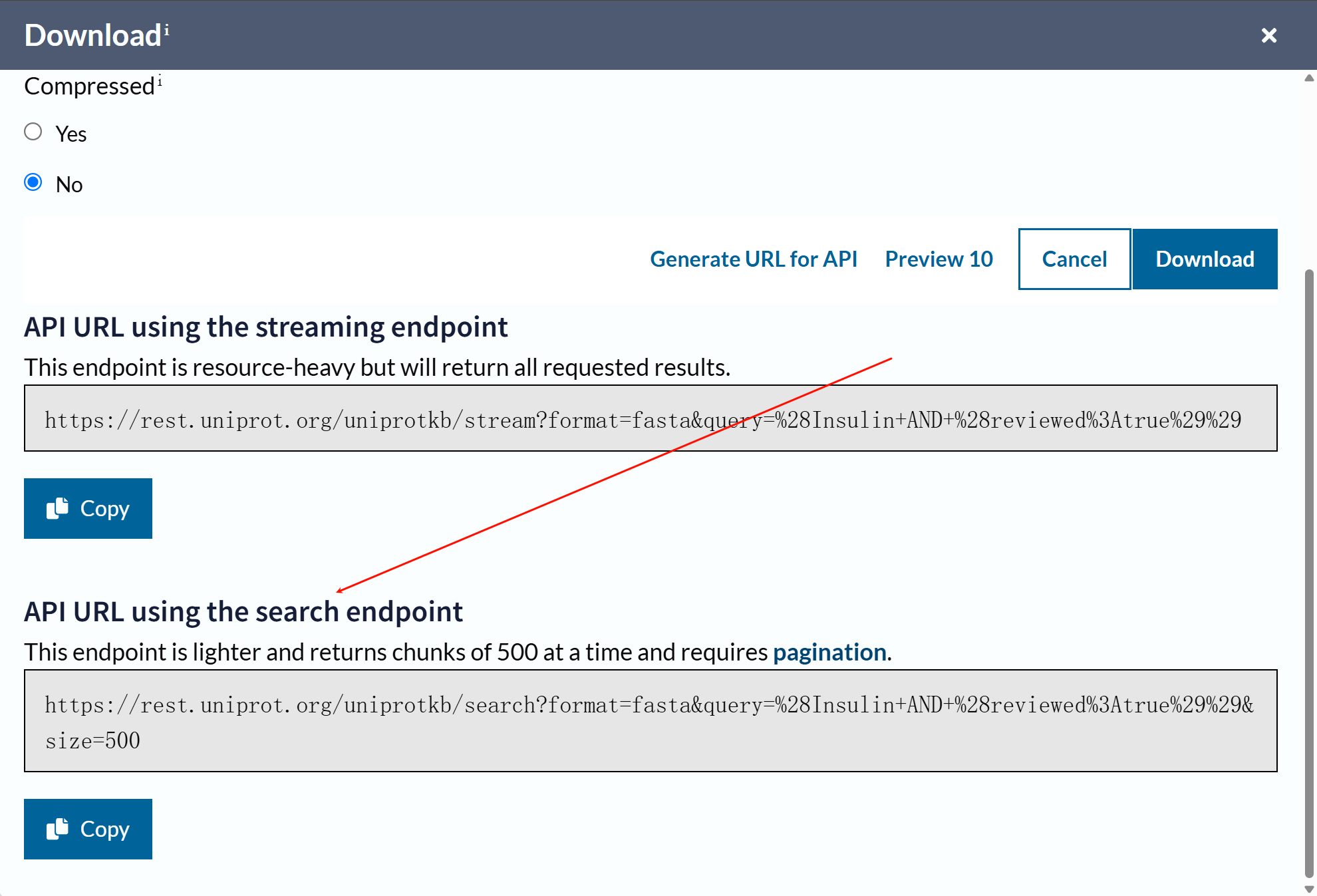
同样的,我们可以使用代码来分析处理数据:
import requests
url = "https://rest.uniprot.org/uniprotkb/search?fields=accession%2Ccc_interaction&format=tsv&query=Insulin%20AND%20%28reviewed%3Atrue%29&size=500"
response = requests.get(url)
print(response)
print(response.text)
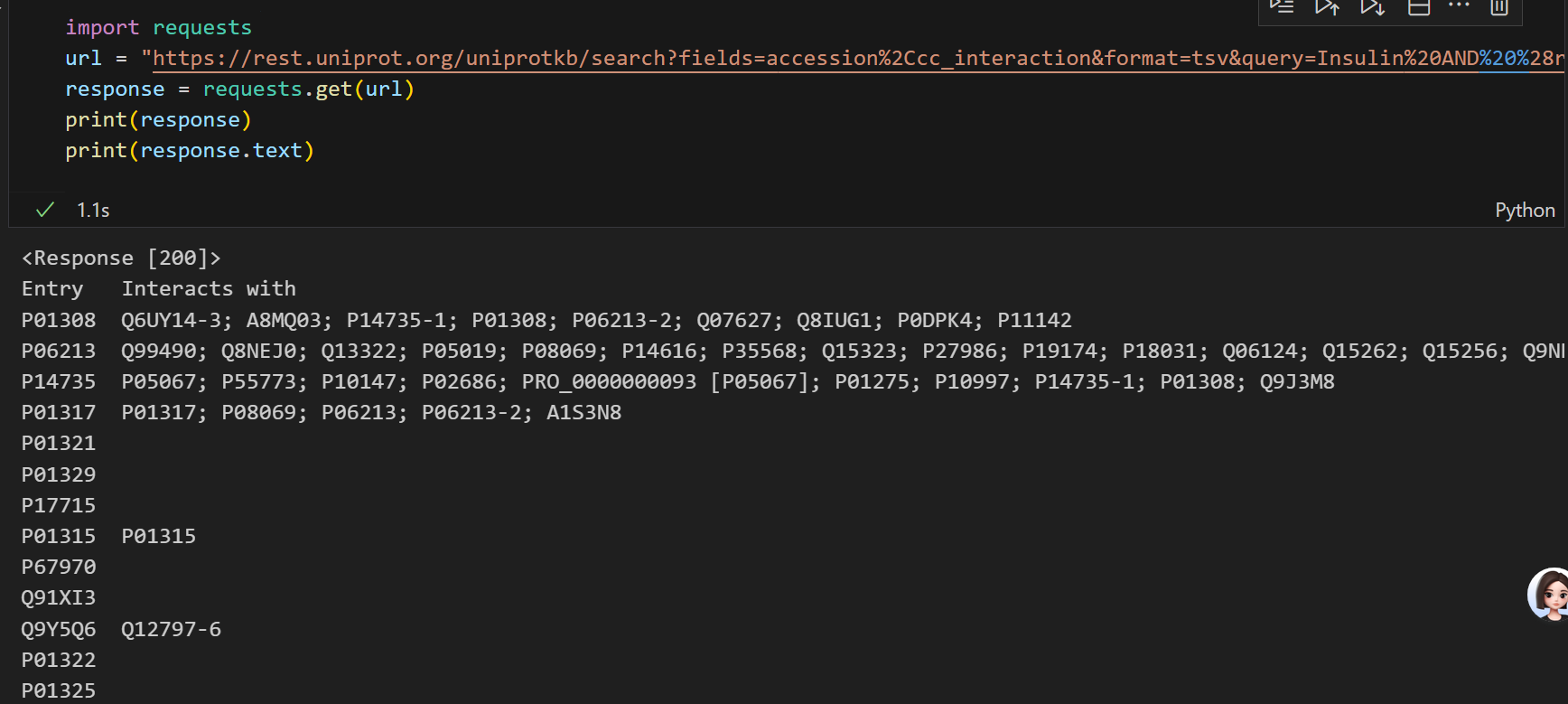
import requests # 发送http请求,调用API
from requests.adapters import HTTPAdapter, Retry # 用于请求重连和连接池管理,增强请求的稳定性(应对网络波动或服务器临时错误)# 正则表达式匹配,提取下一页链接(从uniprot api的分页通过响应头的link字段传递下一页url)
re_next_link = re.compile(r'<(.+)>; rel="next"')
# 重试策略:应对服务器错误
retries = Retry(total=5, # 最大重试次数:5次backoff_factor=0.25, # 重试间隔:每次重试前等待 0.25status_forcelist=[500, 502, 503, 504]) # 仅对“服务器端错误”重试(5xx状态码)
session = requests.Session() # 创建会话(复用连接,提升效率)
session.mount("https://", HTTPAdapter(max_retries=retries)) # 为所有HTTPS请求绑定重试策略# 从响应头获取下一页 URL
def get_next_link(headers):if "Link" in headers: # 检查响应头是否包含“下一页”信息match = re_next_link.match(headers["Link"]) # 用正则匹配URLif match:return match.group(1) # 返回提取的下一页URL,无则返回None# 分页获取数据(生成器函数)
def get_batch(batch_url):while batch_url: # 只要有“下一页”URL,就持续请求response = session.get(batch_url) # 发送GET请求获取当前页数据response.raise_for_status() # 若请求失败(4xx/5xx),直接抛出异常total = response.headers["x-total-results"] # 从响应头获取“总结果数”yield response, total # 用生成器返回“当前页响应”和“总结果数”(避免一次性加载所有数据)batch_url = get_next_link(response.headers) # 更新为下一页URL,循环继续# API 请求 URL
url = 'https://rest.uniprot.org/uniprotkb/search?fields=accession%2Ccc_interaction&format=tsv&query=Insulin%20AND%20%28reviewed%3Atrue%29&size=500'# 存储结果:key=蛋白质编号,value=相互作用数量
interactions = {}
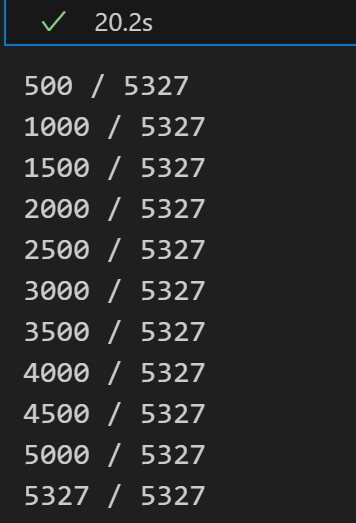
for batch, total in get_batch(url): # 遍历每一页数据for line in batch.text.splitlines()[1:]: # 按行解析TSV:跳过第1行(表头),从第2行开始处理# 拆分“蛋白质编号”和“相互作用列表”primaryAccession, interactsWith = line.split('\t') # 统计相互作用数量:用分号分隔列表,空值则为0interactions[primaryAccession] = len(interactsWith.split(';')) if interactsWith else 0print(f'{len(interactions)} / {total}')
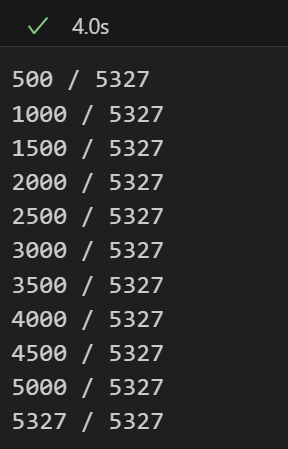
最终interactions字典会存储每个蛋白质编号对应的相互作用数量,便于后续分析(如筛选相互作用最多的蛋白质)。
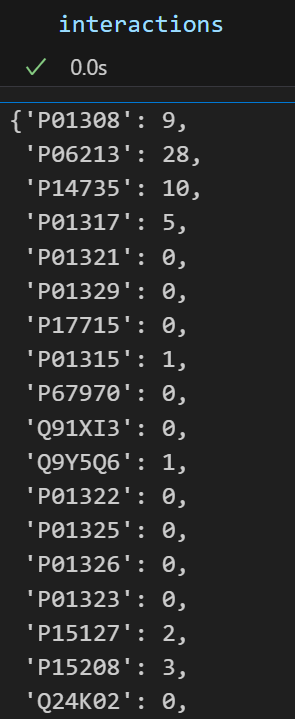
最后,为了查看具有最多相互作用的登录号, 我们可以对interactions字典进行排序:
注意dict对象没有.sort方法

所以我们使用sorted
sorted(interactions.items(), key = lambda x: x[1],reverse=True)
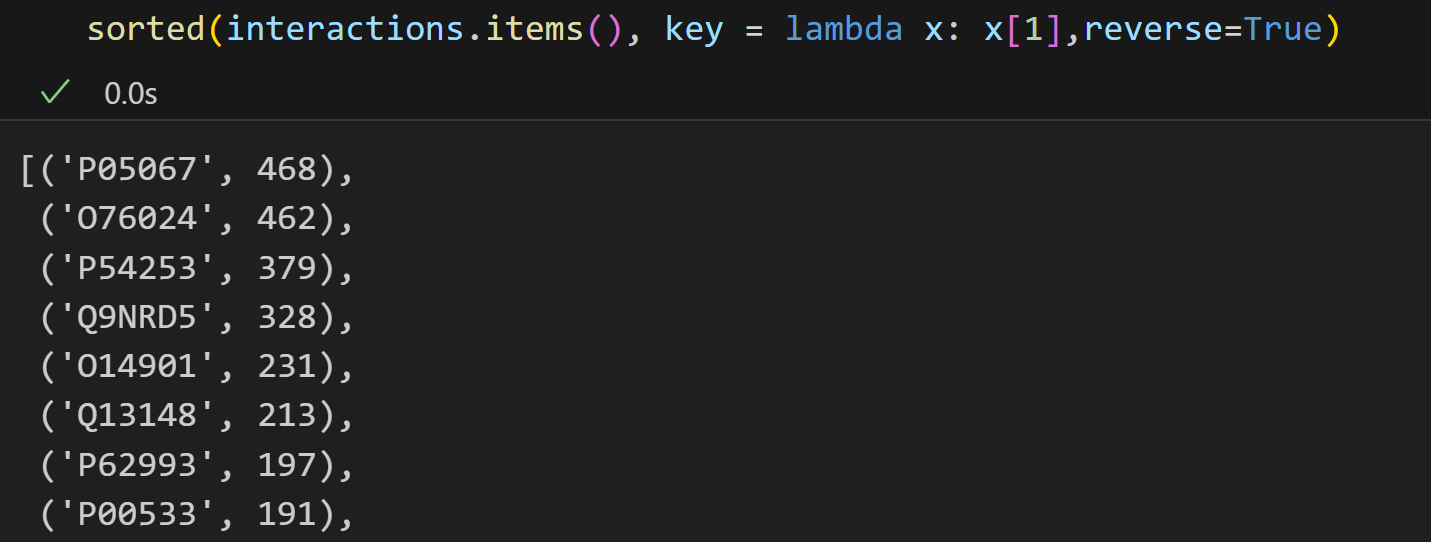
我们可以只看前10个互作多的:
sorted_interactions = sorted(interactions.items(), key=lambda item: item[1], reverse=True)
sorted_interactions[:10]
(5)Python示例—将搜索结果保存到磁盘
除了像前面(4)那样,在python脚本中立即使用搜索结果之外,我们还可以将搜索结果保存起来,比如说保存成一个文件,便于后续随时使用随时分析。
(1)结果数量较少,使用流stream

还是使用前面(4)(1)中对应的例子,
同样在界面操作中获取url,此处略
因为是压缩请求,所以数据都是乱码
可以选择不压缩的url的api,然后简单看一下内容是什么:
import requests
url = 'https://rest.uniprot.org/uniprotkb/stream?compressed=false&format=fasta&query=%28organism_id%3A2697049%29%20AND%20%28reviewed%3Atrue%29'
response = requests.get(url)
print(response.text)
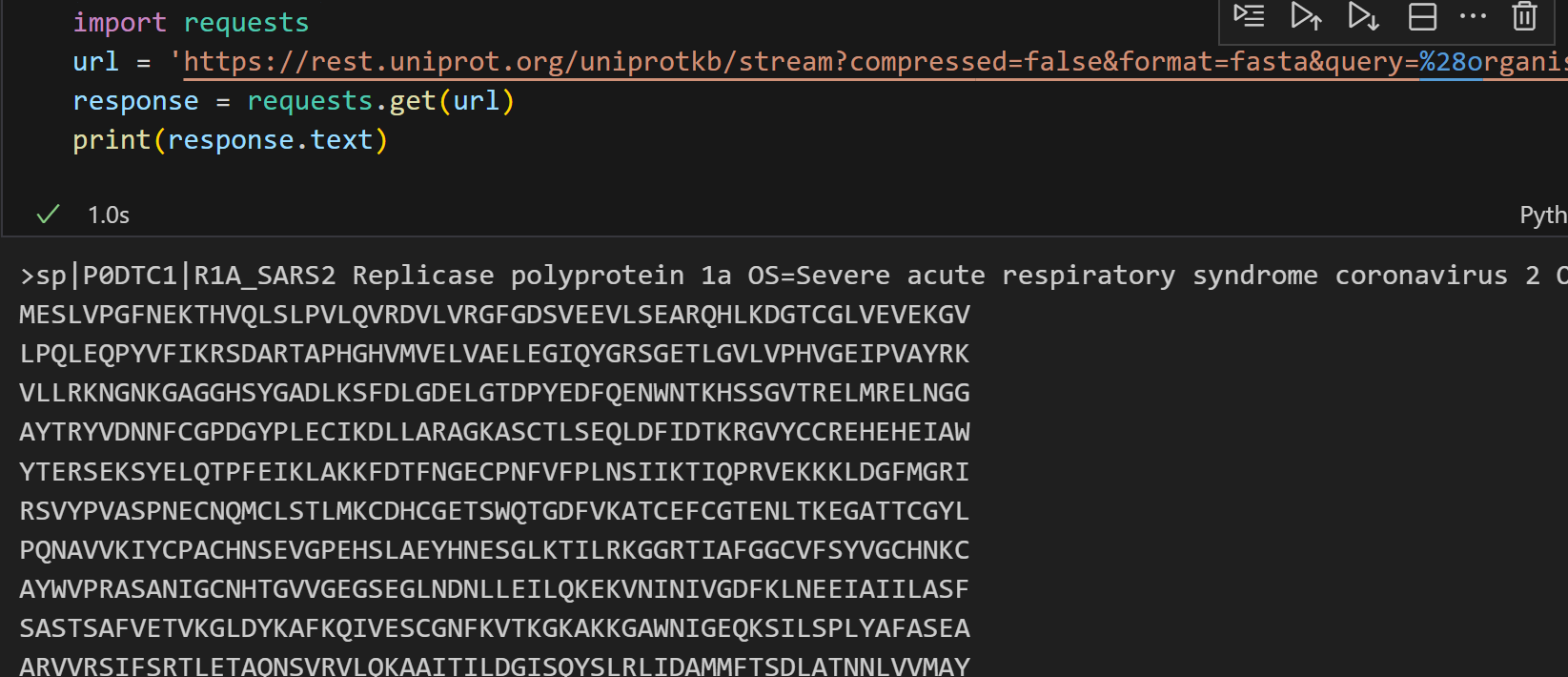
import requests
url = 'https://rest.uniprot.org/uniprotkb/stream?compressed=false&format=fasta&query=%28organism_id%3A2697049%29%20AND%20%28reviewed%3Atrue%29'
with requests.get(url) as response:print(response.text)
总的来说,这就是一个脚本:
通过 HTTP 请求从指定 URL 下载数据,并以二进制流的形式分块写入本地压缩文件
import requestsurl = 'https://rest.uniprot.org/uniprotkb/stream?compressed=true&format=fasta&query=%28organism_id%3A2697049%29%20AND%20%28reviewed%3Atrue%29'
# 向指定的 UniProt API 地址发起 GET 请求,获取数据
with requests.get(url, stream=True) as request:# 关键参数stream=True, 表示以流的形式获取响应内容(逐步获取,而不是一次性获取全部内容)# 若不设此参数,requests 会默认将整个响应内容加载到内存,若文件过大(如超过内存)会导致程序崩溃,适合小文件;stream=True 适合大文件,内存占用极低request.raise_for_status() # 检查请求是否成功,若不成功则抛出异常with open('SARS-CoV-2.fasta.gz', 'wb') as f: # 因为api返回的就是压缩二进制数据, 所以此处需要以二进制模式写入# 返回一个 “迭代器”,每次循环从响应流中取一块数据(chunk),直到所有数据读取完毕# 逐步读取响应流中的数据块# 每次读取的数据块大小为 2^20 字节(即 1MB),可根据需要调整 chunk_size 的值for chunk in request.iter_content(chunk_size=2**20):# 将每次读取的二进制数据块(chunk)写入到本地文件 f 中f.write(chunk)
数据获取完毕,确实效果如下:
我们可以实际稍微查看一下这个压缩文件,
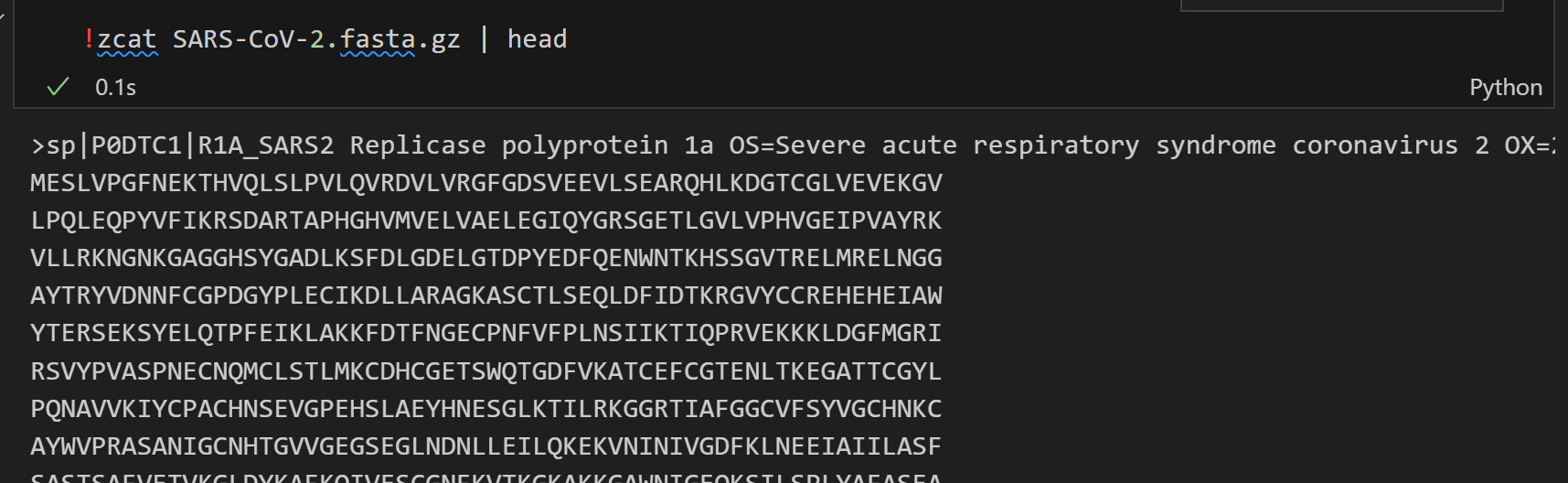
!zcat SARS-CoV-2.fasta.gz | head
或者使用gzcat
(2)结果数量较多,使用分页pagination
同样使用前面(4)(2)的例子,同样获取url API
import re # 正则表达式匹配
import requests # 发送http请求, 调用API
from requests.adapters import HTTPAdapter, Retry # 用于请求重连和连接池管理re_next_link = re.compile(r'<(.+)>; rel="next"') # 正则表达式, 用于匹配下一页的链接(uniprot api的响应headers中,会通过link字段返回分页链接)。从Link字段中提取出下一页的API地址# 请求重试策略, 遇到500, 502, 503, 504状态码时重试5次, 每次重试间隔0.25秒
# API 偶尔会返回 5xx 错误(服务器过载、临时故障),通过重试避免单次失败导致程序中断
retries = Retry(total=5, # 最大重试次数5次backoff_factor=0.25, # 重试间隔,每次重试前等待0.25秒status_forcelist=[500, 502, 503, 504]) # 需要重试的状态码列表(仅对“服务器错误”重试(5xx状态码))
session = requests.Session() # 创建会话(复用连接,提升效率)
session.mount("https://", HTTPAdapter(max_retries=retries)) # 为所有HTTPS请求绑定重试策略# 从响应头提取下一页url
def get_next_link(headers):if "Link" in headers:match = re_next_link.match(headers["Link"])if match:return match.group(1)# 分页获取数据(生成器函数)
def get_batch(batch_url):while batch_url:response = session.get(batch_url)response.raise_for_status()total = response.headers["x-total-results"]yield response, totalbatch_url = get_next_link(response.headers)# 下面是主程序
# API请求参数
url = 'https://rest.uniprot.org/uniprotkb/search?fields=accession%2Ccc_interaction&format=tsv&query=Insulin%20AND%20%28reviewed%3Atrue%29&size=500'
# 分页保存数据+显示进度
progress = 0
with open('insulin-interactions.tsv', 'w') as f:for batch, total in get_batch(url):lines = batch.text.splitlines()if not progress:print(lines[0], file=f)for line in lines[1:]:print(line, file=f)progress += len(lines[1:])print(f'{progress} / {total}')
同样,结果数据如下:
3,Query/Return fields 查询/返回字段

查询字段,参考链接:https://www.uniprot.org/help/query-fields
返回字段,参考链接:https://www.uniprot.org/help/return_fields

外部引用数据库,参考链接:https://www.uniprot.org/help/return_fields_databases
4,其他
(1)不同数据库之间的ID映射(mapping)

参考官方链接:https://www.uniprot.org/help/id_mapping
除了主界面的交换工具外,
ID映射也支持程序化访问:

程序化映射,参考链接:https://www.uniprot.org/help/id_mapping_prog
https://www.uniprot.org/api-documentation/idmapping
(2)REST API 中的 HTTP 状态码
参考链接:https://www.uniprot.org/help/rest-api-headers
5,两个简单的脚本示例
(1)没有蛋白质id,想找到符合query条件的蛋白质id(及其数据)
重点是找到id,有了id,其实序列、其他unirprot中注释的属性就都很简单了。
此处主要是参考前面2(通过query查询检索条目)。
还是以CTCF为例子,数据条目见https://www.uniprot.org/uniprotkb/P49711/entry#family_and_domains,

这里我随便举一个例子,比如说我想要这两个数据库引用都有的(OR关系)的蛋白质条目,
我可以在搜索界面中这么写:
organism_id:9606 AND reviewed:true AND (xref:prosite-PS00028 OR xref:prosite-PS50157)
注意,在界面搜索的时候,不要将query写成下面这样,就是在搜索交互界面上不要写“+”,这个和程序逻辑是不一样的。
(organism_id:9606)+AND+(reviewed:true)+AND+((xref:prosite-PS00028)+OR+(xref:prosite-PS50157))
前者的逻辑搜索结果是:791条
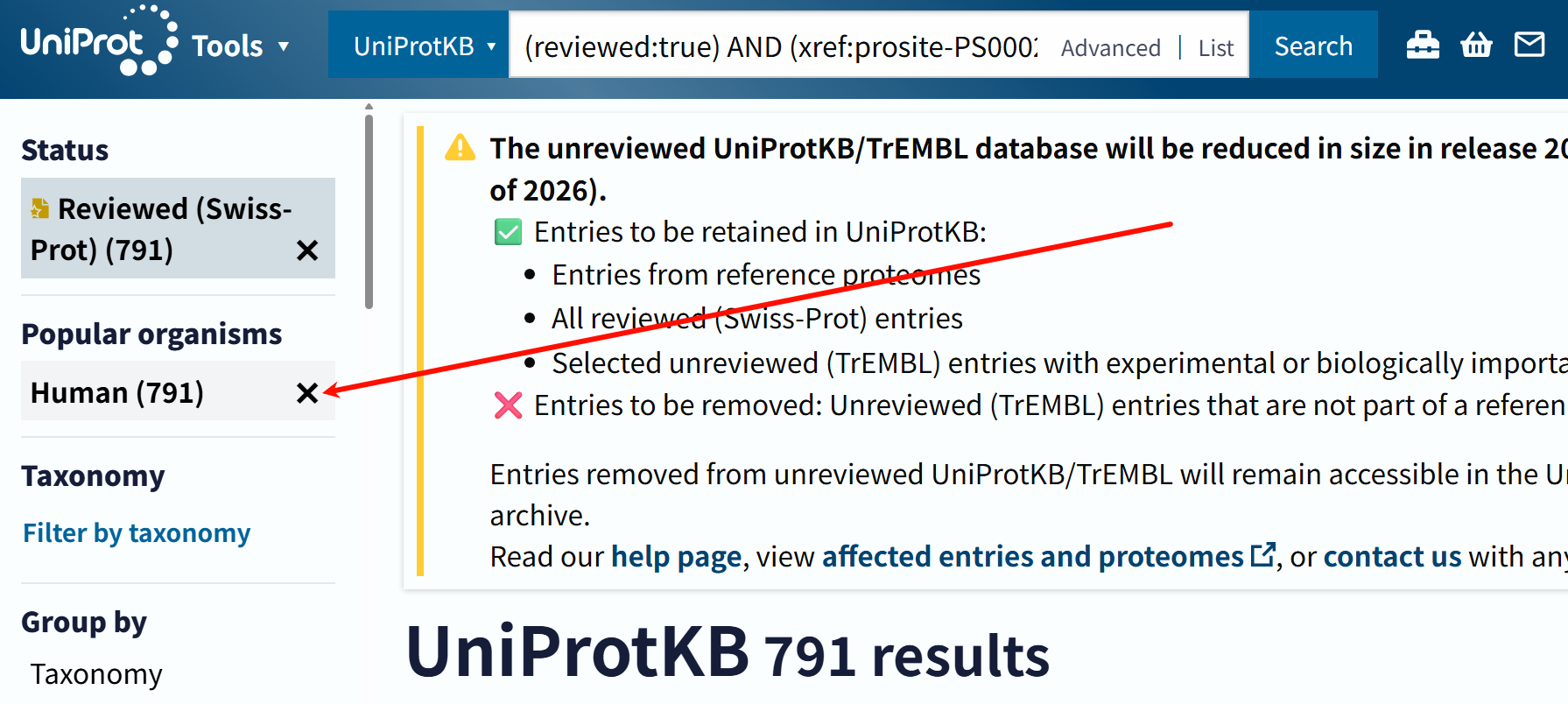
后者的搜索结果就是:728条
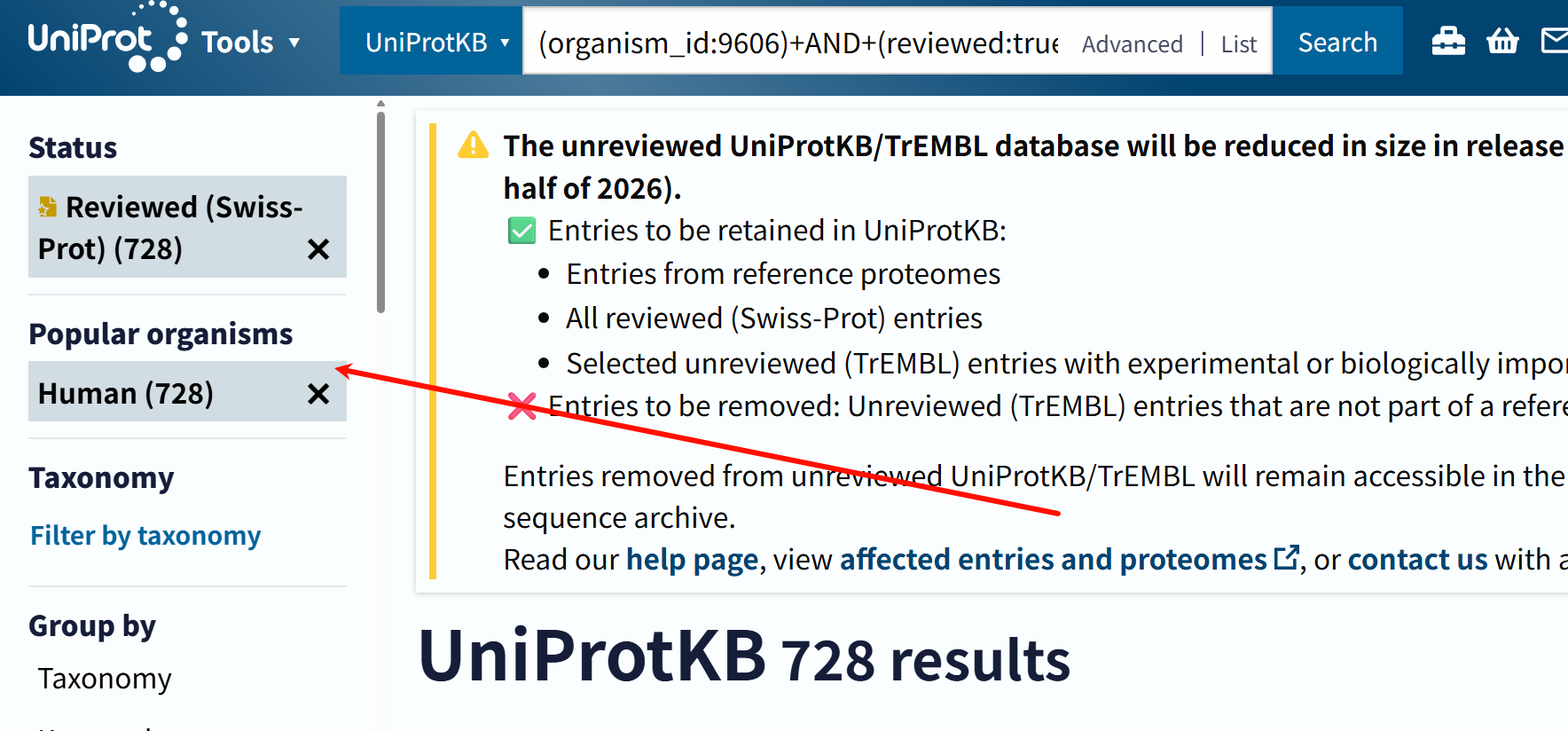
就是因为在搜索界面中多写了个“+”号,搜索结果就会大大减少。
好,现在我们来看一下按照这样的query,构造的格式,在程序中会返回什么样的结果。
import requests
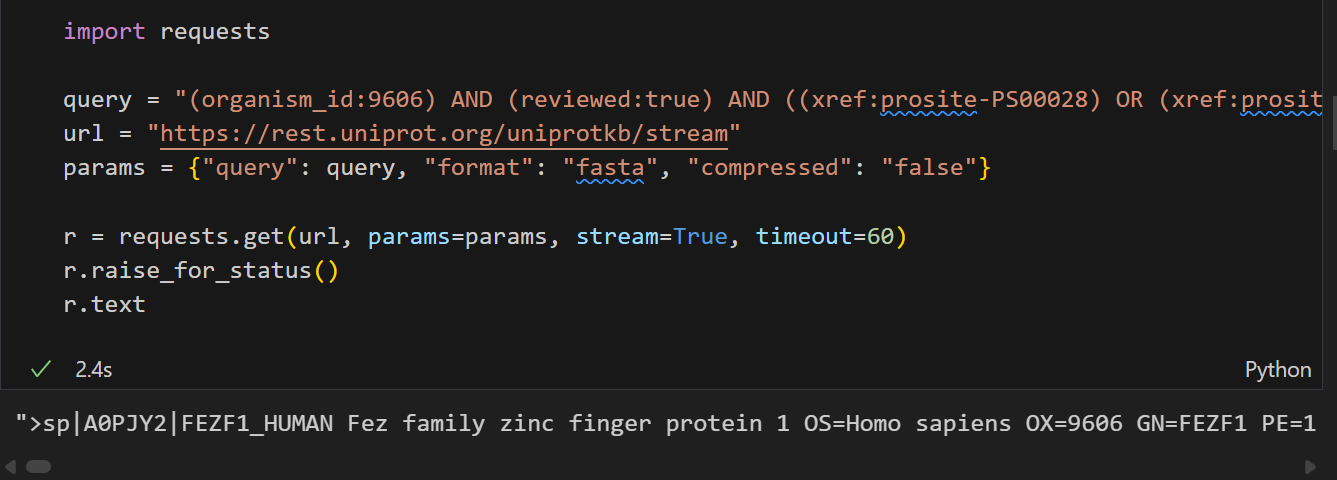
url = "https://rest.uniprot.org/uniprotkb/search?query=(organism_id:9606)+AND+(reviewed:true)+AND+(xref:prosite-PS00028)&format=fasta&compressed=false"
response = requests.get(url)
if response.status_code == 200:fasta_data = response.textprint(fasta_data)
else:print(f"Failed to fetch data. Status code: {response.status_code}")
看起来返回了我们想要的条目数据,这应该是一个好消息!
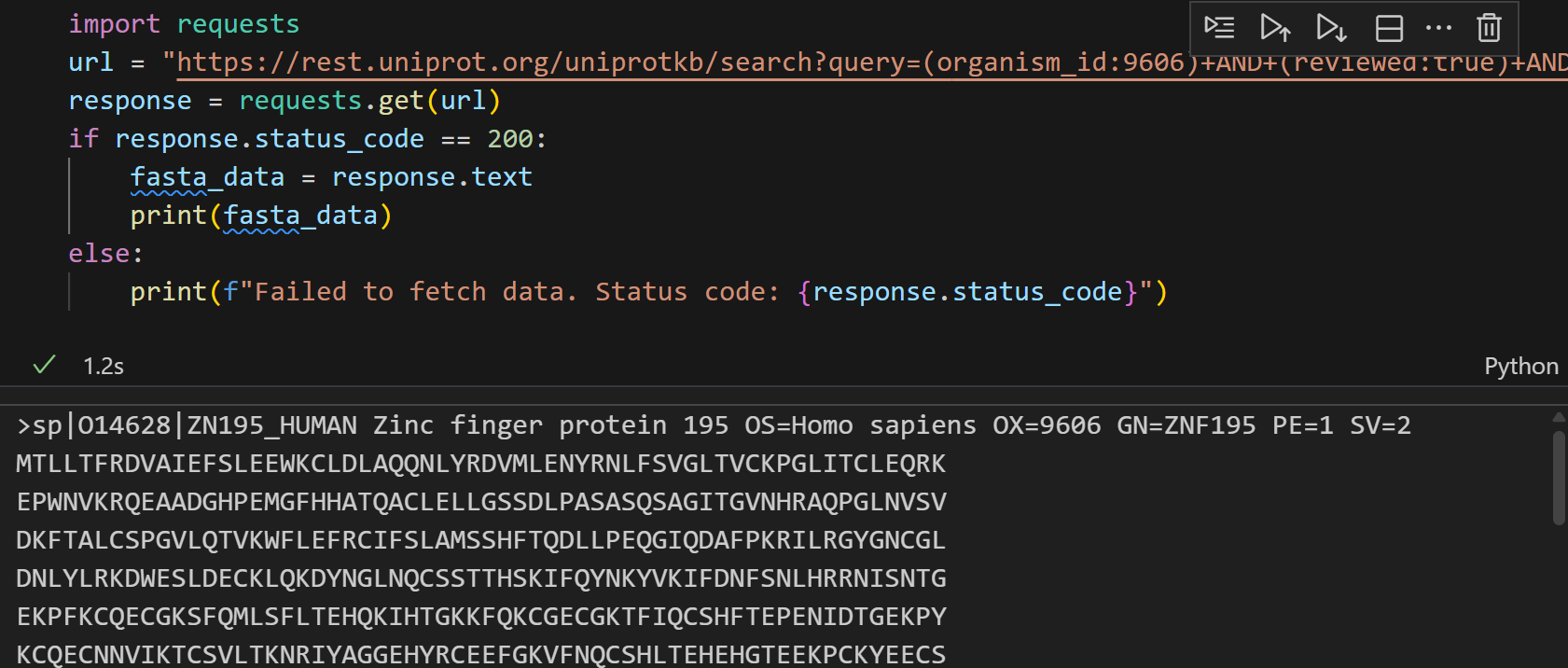
但是实际检查发现,数目太少了!完全对应不上!
这是因为分页的问题:UniProt REST /uniprotkb/search 默认每页只有少量结果,
需要自己翻页,否则就只能看到第一页。
解决方案也很简单,
- 提高每页大小并用cursor翻页
简单草稿代码如下,
import requests, time
def get_Uniprot_protein_fasta(query: str, format: str, compressed: bool, size: int) -> list[str]:"""Description:根据给定的查询参数, 从Uniprot数据库中获取蛋白质的fasta序列, 支持分页请求;Args:query (str): 查询字符串, 用于指定搜索条件, 可以是单个序列数据, 也可以是批量查询;format (str): 返回数据的格式, 默认为"fasta";compressed (bool): 是否请求压缩格式的数据, 默认为False;size (int): 每页返回的记录数/单次请求返回的序列数, 默认为500Returns:list[str]: 包含所有获取到的fasta序列的列表"""base = "https://rest.uniprot.org/uniprotkb/search"params = {"query": query,"format": format,"compressed": compressed,"size": size}# 用于存储分页请求返回的fasta序列片段fasta_parts = []url = base# 分页请求循环,直到没有下一页while True:# 首次请求时使用params字典,后续请求的"下一页"url已包含所有必要参数,因此不再重复传入params参数# 请求超时设置为60秒r = requests.get(url, params=params if url == base else None, timeout=60)# 检查是否请求成功,否则抛出异常r.raise_for_status()# 解析响应内容,将当前请求返回的fasta序列添加到列表中fasta_parts.append(r.text)# 解析下一页url# 先尝试获取 “next” 对应的子字典,若不存在则返回空字典;再从子字典中获取 “url”,若不存在则返回None(表示无下一页)next_url = r.links.get("next", {}).get("url")# 若无下一页,则跳出循环if not next_url:break# 更新url为下一页的url,继续请求# next_url已包含所有参数,无需重复传递url, params = next_url, None# 为避免过于频繁请求,稍作延时time.sleep(0.2)# 返回所有获取到的fasta序列片段, 拼接为一个完整的字符串fasta_str = "".join(fasta_parts)# 最好分割为一个accession对应一个字符串的列表返回fasta_list = fasta_str.strip().split("\n>")return fasta_list
我们还是使用P49711为例,作为query测试,
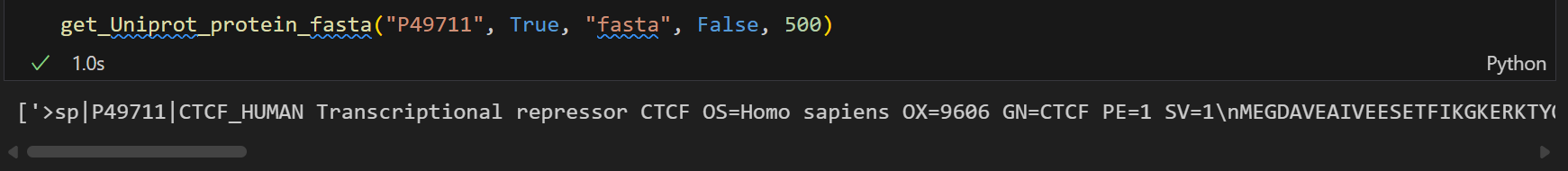
所以accession可以作为一个query,进行查询获取数据。
但是奇怪的是,如果我们把reviewed也作为query中的内容,
因为按照具体的query fields参考https://www.uniprot.org/help/query-fields,
reviewed其实也作为query语法中的一部分:

如果我们写成
import requests, time
def get_Uniprot_protein_fasta(query: str, format: str, compressed: bool, size: int) -> list[str]:"""Description:根据给定的查询参数, 从Uniprot数据库中获取蛋白质的fasta序列, 支持分页请求;Args:query (str): 查询字符串, 用于指定搜索条件, 可以是单个序列数据, 也可以是批量查询;format (str): 返回数据的格式, 默认为"fasta";compressed (bool): 是否请求压缩格式的数据, 默认为False;size (int): 每页返回的记录数/单次请求返回的序列数, 默认为500Returns:list[str]: 包含所有获取到的fasta序列的列表"""base = "https://rest.uniprot.org/uniprotkb/search"params = {"query": query,"format": format,"compressed": compressed,"size": size}# 用于存储分页请求返回的fasta序列片段fasta_parts = []url = base# 分页请求循环,直到没有下一页while True:# 首次请求时使用params字典,后续请求的"下一页"url已包含所有必要参数,因此不再重复传入params参数# 请求超时设置为60秒r = requests.get(url, params=params if url == base else None, timeout=60)# 检查是否请求成功,否则抛出异常r.raise_for_status()# 解析响应内容,将当前请求返回的fasta序列添加到列表中fasta_parts.append(r.text)# 解析下一页url# 先尝试获取 “next” 对应的子字典,若不存在则返回空字典;再从子字典中获取 “url”,若不存在则返回None(表示无下一页)next_url = r.links.get("next", {}).get("url")# 若无下一页,则跳出循环if not next_url:break# 更新url为下一页的url,继续请求# next_url已包含所有参数,无需重复传递url, params = next_url, None# 为避免过于频繁请求,稍作延时time.sleep(0.2)# 返回所有获取到的fasta序列片段, 拼接为一个完整的字符串fasta_str = "".join(fasta_parts)# 最好分割为一个accession对应一个字符串的列表返回fasta_list = fasta_str.strip().split("\n>")return fasta_list
get_Uniprot_protein_fasta("P49711 and reviewed:true", "fasta", False, 500)
那么结果就大不一样了:
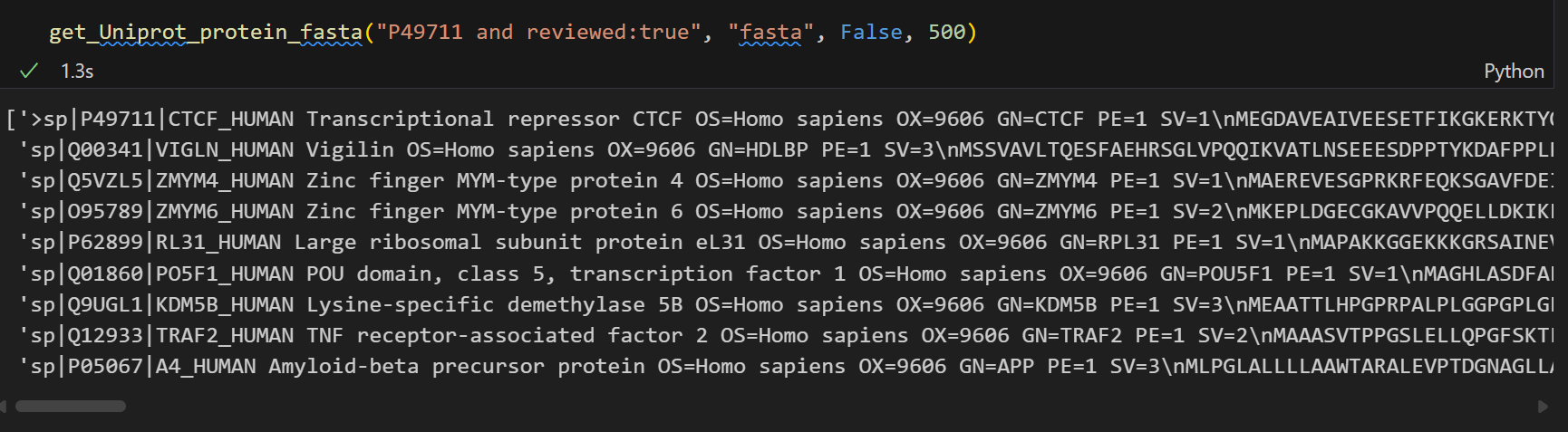
也就是说:
下面两个获得的结果是不一样的
# reviewed放在query中
get_Uniprot_protein_fasta("P49711 and reviewed:true", "fasta", False, 500)# reviewed放在params参数中
get_Uniprot_protein_fasta("P49711", True, "fasta", False, 500)为了解决上面这个疑问,我们这里做一个测试,打印请求得到的url,来看看到底有什么区别:
import requests, time
def get_Uniprot_protein_fasta(query: str, format: str, compressed: bool, size: int) -> list[str]:"""Description:根据给定的查询参数, 从Uniprot数据库中获取蛋白质的fasta序列, 支持分页请求;Args:query (str): 查询字符串, 用于指定搜索条件, 可以是单个序列数据, 也可以是批量查询;format (str): 返回数据的格式, 默认为"fasta";compressed (bool): 是否请求压缩格式的数据, 默认为False;size (int): 每页返回的记录数/单次请求返回的序列数, 默认为500Returns:list[str]: 包含所有获取到的fasta序列的列表"""base = "https://rest.uniprot.org/uniprotkb/search"params = {"query": query,"format": format,"compressed": compressed,"size": size}# 用于存储分页请求返回的fasta序列片段fasta_parts = []url = base# 分页请求循环,直到没有下一页while True:# 首次请求时使用params字典,后续请求的"下一页"url已包含所有必要参数,因此不再重复传入params参数# 请求超时设置为60秒r = requests.get(url, params=params if url == base else None, timeout=60)# 检查是否请求成功,否则抛出异常r.raise_for_status()# 打印观察当前请求的url(可选)print(f"Requesting URL: {r.url}")# 解析响应内容,将当前请求返回的fasta序列添加到列表中# 新加的!!!fasta_parts.append(r.text)# 解析下一页url# 先尝试获取 “next” 对应的子字典,若不存在则返回空字典;再从子字典中获取 “url”,若不存在则返回None(表示无下一页)next_url = r.links.get("next", {}).get("url")# 若无下一页,则跳出循环if not next_url:break# 更新url为下一页的url,继续请求# next_url已包含所有参数,无需重复传递url, params = next_url, None# 为避免过于频繁请求,稍作延时time.sleep(0.2)# 返回所有获取到的fasta序列片段, 拼接为一个完整的字符串fasta_str = "".join(fasta_parts)# 最好分割为一个accession对应一个字符串的列表返回fasta_list = fasta_str.strip().split("\n>")return fasta_list # 测试!!!
get_Uniprot_protein_fasta("P49711 and reviewed:true", "fasta", False, 500)
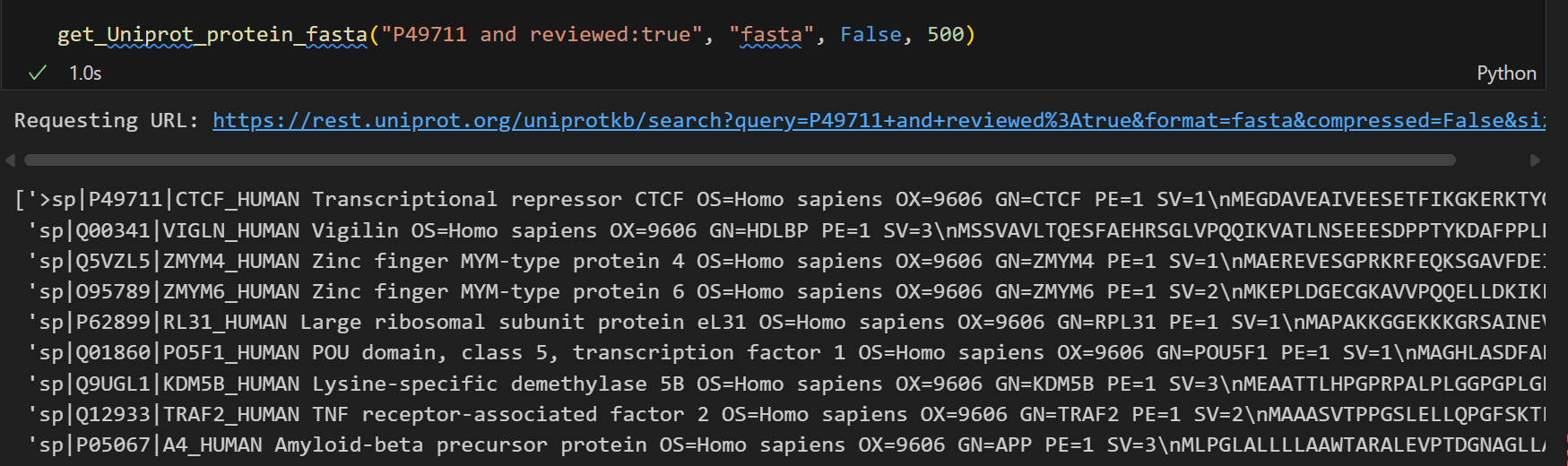
可以看到,我们获得的url是
https://rest.uniprot.org/uniprotkb/search?query=P49711+and+reviewed%3Atrue&format=fasta&compressed=False&size=500
打开这个链接,确实是有很多条entry,不仅仅是P49711
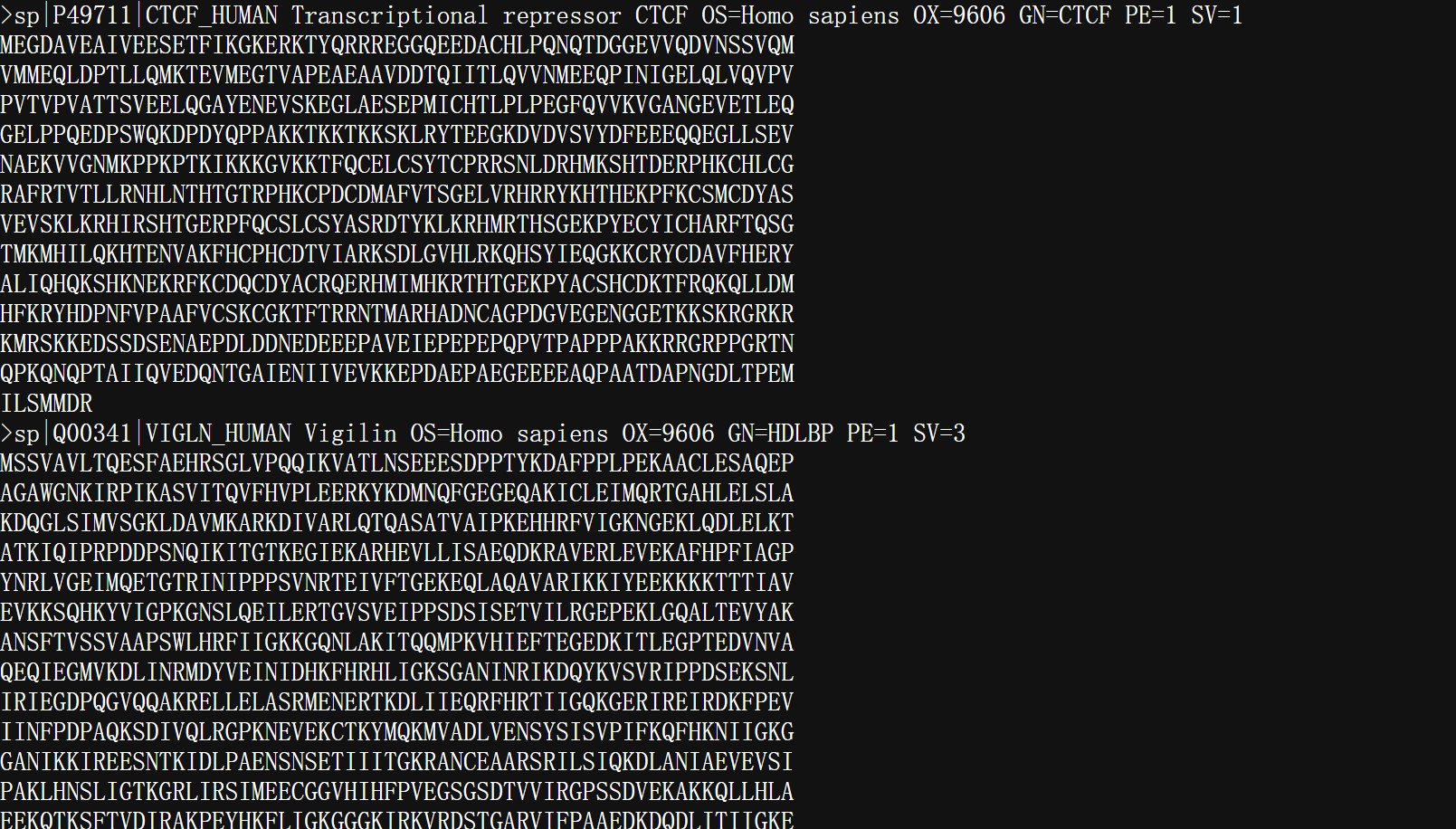
这说明这个结果确实没有返回错误,
有问题的是我们对于这个query的理解,最简单直接的方法就是返回去查看这个query的检索结果是什么,直接在uniprot搜索框中输入:
一字不差,但是确实也是输出9条,是不是很奇怪,明明我指定了P49711作为唯一的accession?
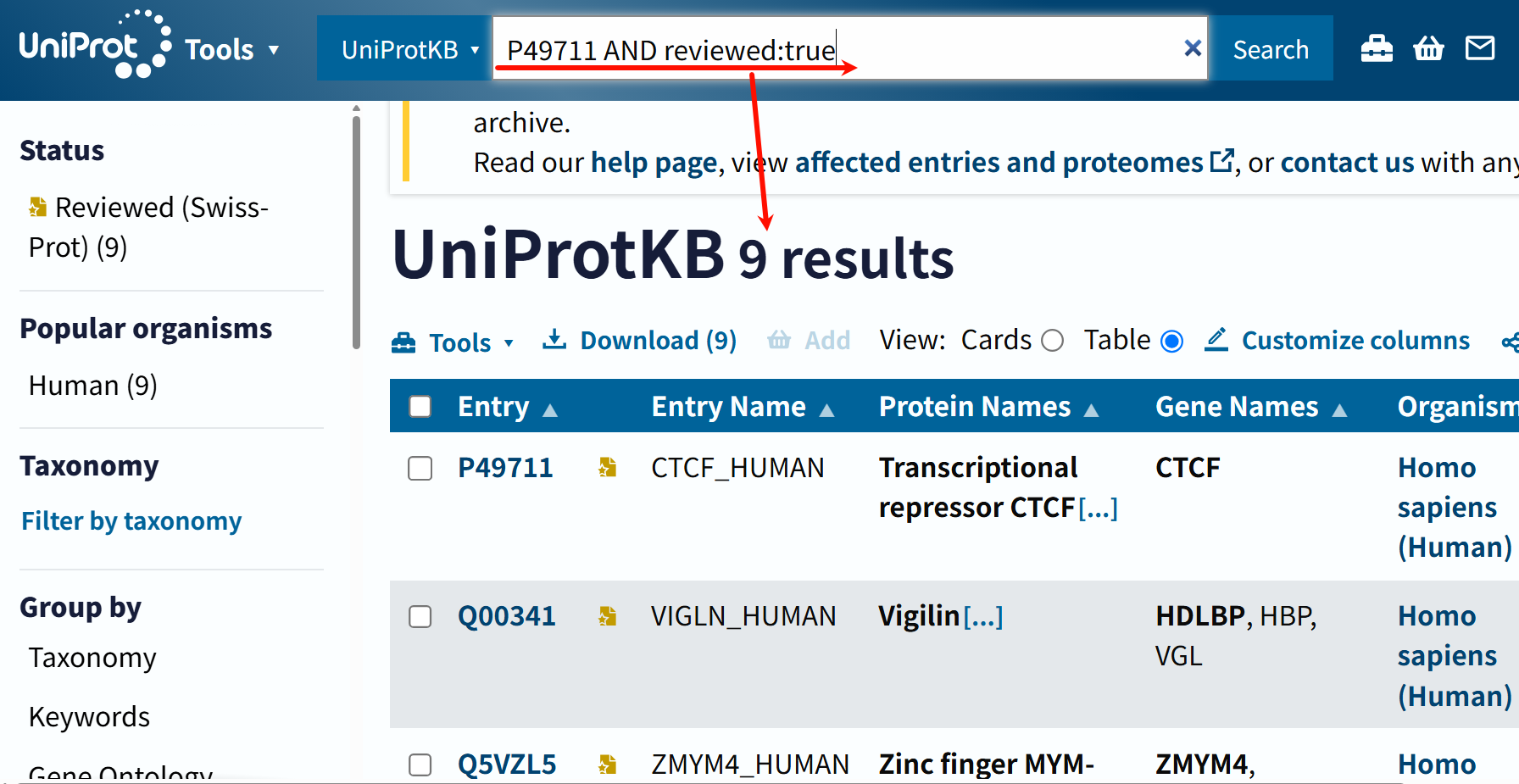
只可能有一个问题,就是对于query中的accession,其实我们也是需要按照一定格式来写的:
按理说单个条目的搜索,其实最好是增加accesion的field进行,

其实最好是使用下面的这个,因为下面的这个是使用primary ac:


总得来说,因为我的数据中,没有isoform(我暂时不考虑),所以我不会动isoform的url参数,那么用accession和accession_id对于我来说结果其实是一样的。
我们可以在url的格式中用includeisoform,
参考:https://www.uniprot.org/help/api_queries

requests.get("https://rest.uniprot.org/uniprotkb/search?query=accession:P49711 and reviewed:true&format=fasta&compressed=false&size=500&includeIsoform=true").text.split("\n>")

对照网页端,确实CTCF有两个异构体。
我们再测试一下,
get_Uniprot_protein_fasta("accession:P49711 and reviewed:true", "fasta", False, 500)
确实:效果和返回的url都是唯一的了
返回的url打开也只有一个:

get_Uniprot_protein_fasta("accession_id:P49711 and reviewed:true", "fasta", False, 500)
当然,上面只是一个简单的草稿脚本,实际的params参数字典我们都可以自定义再传入,在一次查询中只传入query和parms_dict两个参数,然然后再进行合并,合并到url的http请求的参数中。
import requests, time
def get_Uniprot_protein_fasta(query: str, params_dict: dict) -> list[str]:"""Description:根据给定的查询参数, 从Uniprot数据库中获取蛋白质的fasta序列, 支持分页请求;Args:query (str): 查询字符串, 用于指定搜索条件, 可以是单个序列数据,也可以是批量查询;params_dict (dict): 其他请求参数的字典, 一般包括以下键值对:format (str): 返回数据的格式, 默认为"fasta";compressed (bool): 是否请求压缩格式的数据, 默认为False;size (int): 每页返回的记录数/单次请求返回的序列数, 默认为500Returns:list[str]: 包含所有获取到的fasta序列的列表Notes:1, params_dict中的键值对会被添加到请求参数中, query参数会单独传递, 虽然参数单独传递, 但是最后都合并到URL中进行请求。2, query参数参考https://www.uniprot.org/help/query-fields, params_dict参数则比较固定, 参考Args部分说明;3, query参数一般建议带上物种, organism_id:9606; 以及是否reviewed:true等过滤条件"""base = "https://rest.uniprot.org/uniprotkb/search"# 用于存储分页请求返回的fasta序列片段fasta_parts = []url = base# 将查询参数添加到params_dict中params_dict["query"] = query# 分页请求循环,直到没有下一页while True:# 首次请求时使用params字典,后续请求的"下一页"url已包含所有必要参数,因此不再重复传入params参数# 请求超时设置为60秒r = requests.get(url, params=params_dict if url == base else None, timeout=60)# 检查是否请求成功,否则抛出异常r.raise_for_status()# 解析响应内容,将当前请求返回的fasta序列添加到列表中fasta_parts.append(r.text)# 解析下一页url# 先尝试获取 “next” 对应的子字典,若不存在则返回空字典;再从子字典中获取 “url”,若不存在则返回None(表示无下一页)next_url = r.links.get("next", {}).get("url")# 若无下一页,则跳出循环if not next_url:break# 更新url为下一页的url,继续请求# next_url已包含所有参数,无需重复传递url = next_url# 为避免过于频繁请求,稍作延时time.sleep(0.2)# 返回所有获取到的fasta序列片段, 拼接为一个完整的字符串fasta_str = "".join(fasta_parts)# 最好分割为一个accession对应一个字符串的列表返回fasta_list = fasta_str.strip().split("\n>")return fasta_list
单个条目的查询效果如下:
query = "accession:P49711 AND reviewed:true"
params_dict = {"format": "fasta","compressed": False,"size": 500
}
get_Uniprot_protein_fasta(query=query,params_dict=params_dict)
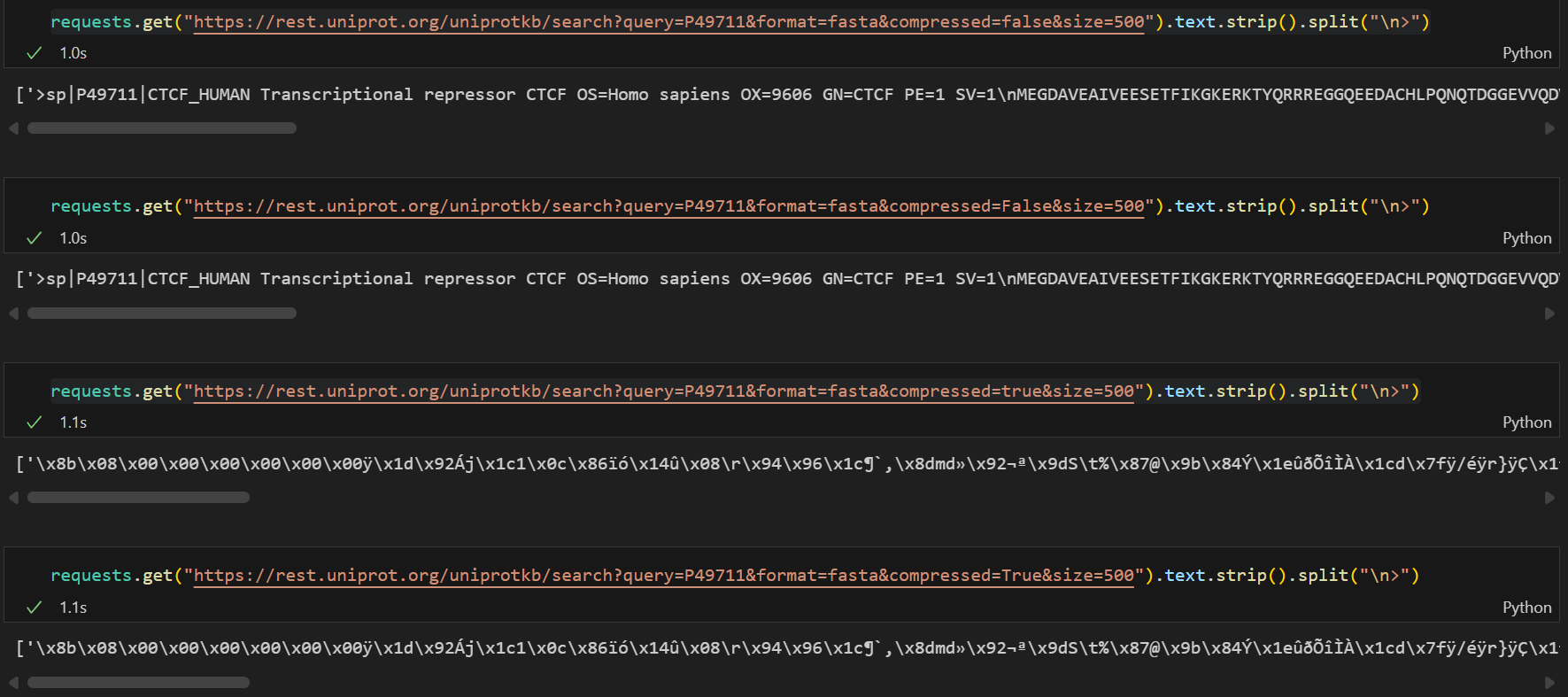
另外我测试过程中发现的一点是:
在http的请求中,query中涉及到bool值的,其实标注为str,
也就是true/True,false/False,实际效果上并没有区别

当然,这里其实对应前面检索单个条目+reviewed时,要不要对单个条目加accession的问题:
我发现,其实如果直接在query中搜索单个item的ac,不加上什么其他乱七八糟的query关键词,比如说reviewed,其实单个条目也不需要加accession限制词:
直接结果就是一个:其实这就和我们在uniprot网页端界面中直接搜索id ac号是一样的逻辑和返回结果

当然,鉴于我习惯在query中加上reviewed关键词,
所以为了我单个条目搜索和多个条目搜索都能够正常执行,其实还是要规范写法,都加上accession词。
好,现在回到我们前面的问题,我们直接搜索是少791条entry,
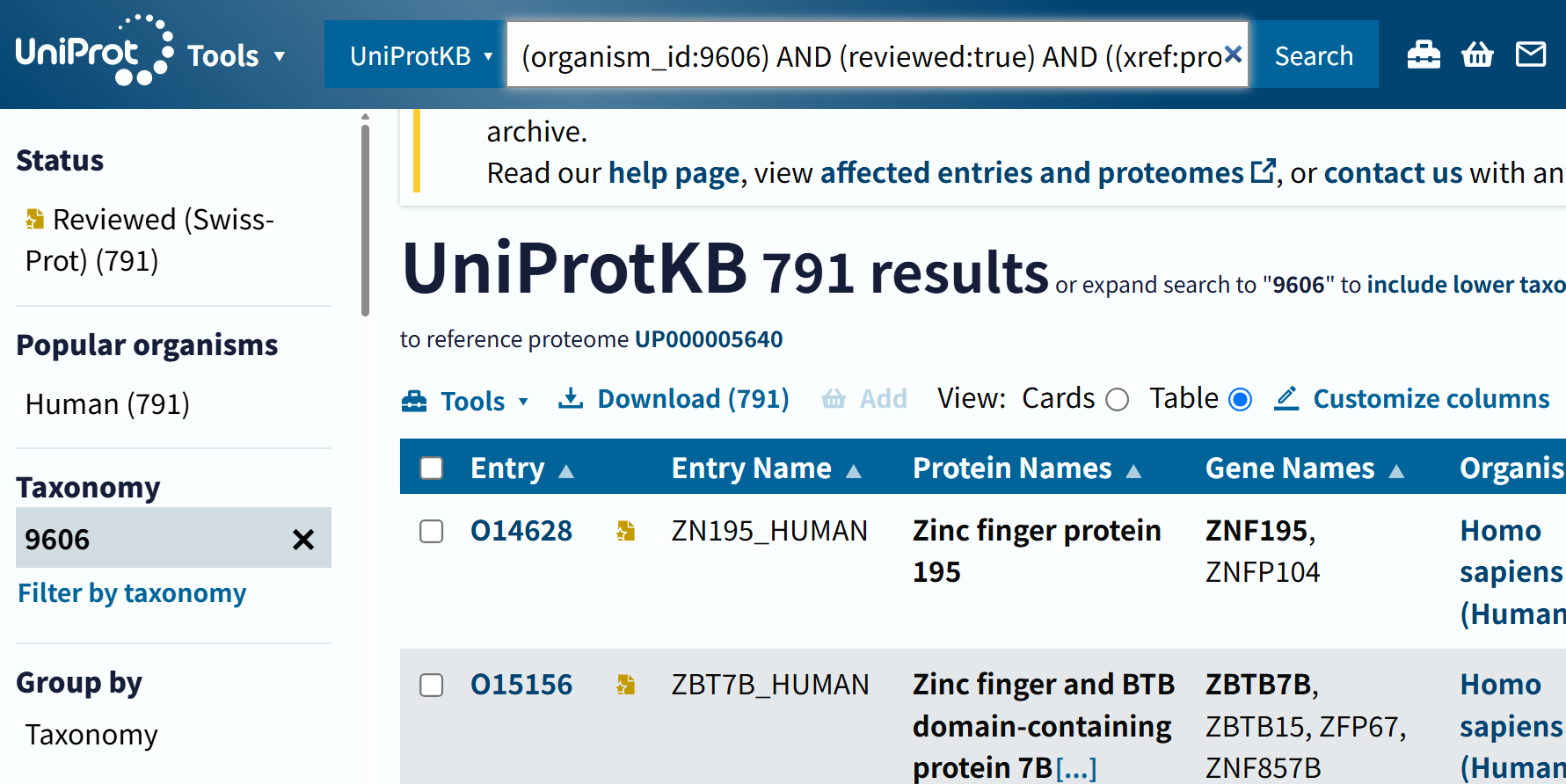
修复之后的代码部分:
query = "(organism_id:9606) AND (reviewed:true) AND ((xref:prosite-PS00028) OR (xref:prosite-PS50157))"
params_dict = {"format": "fasta","compressed": False,"size": 500
}results = get_Uniprot_protein_fasta(query=query, params_dict=params_dict)
len(results)
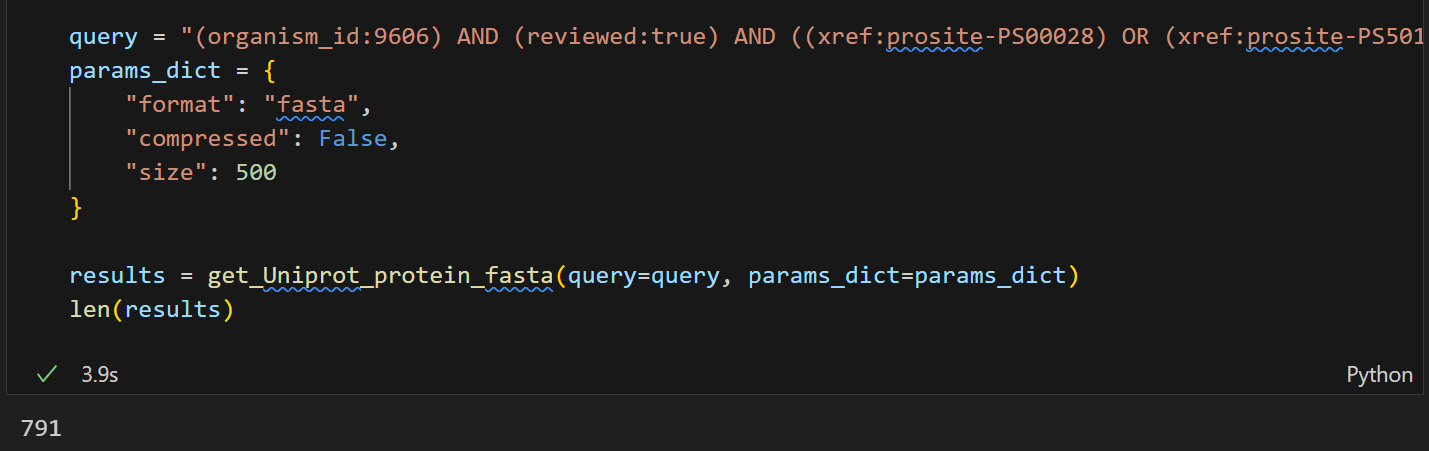
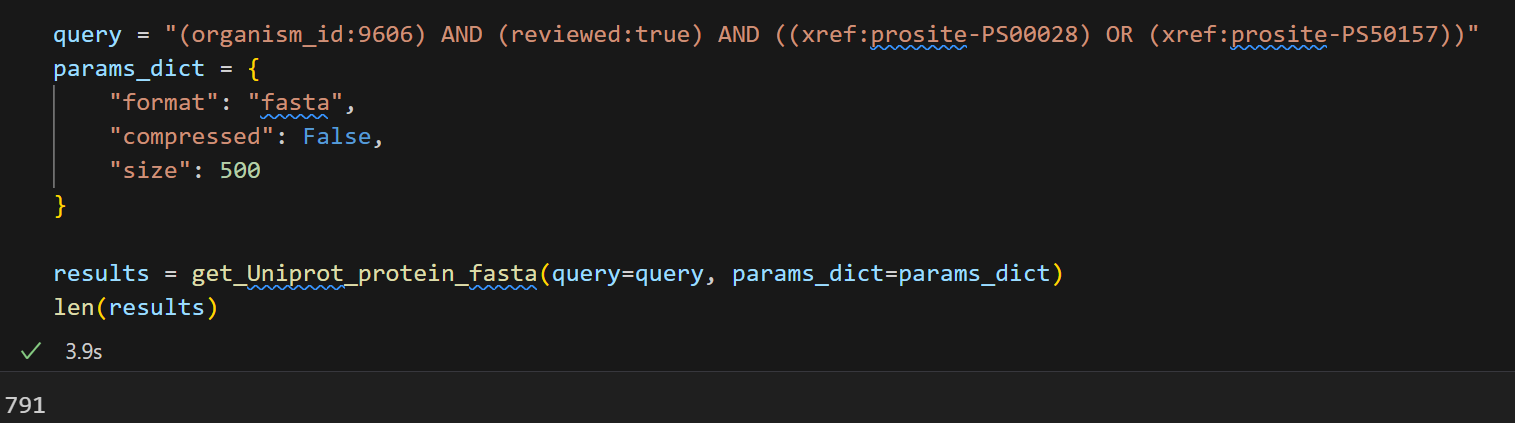
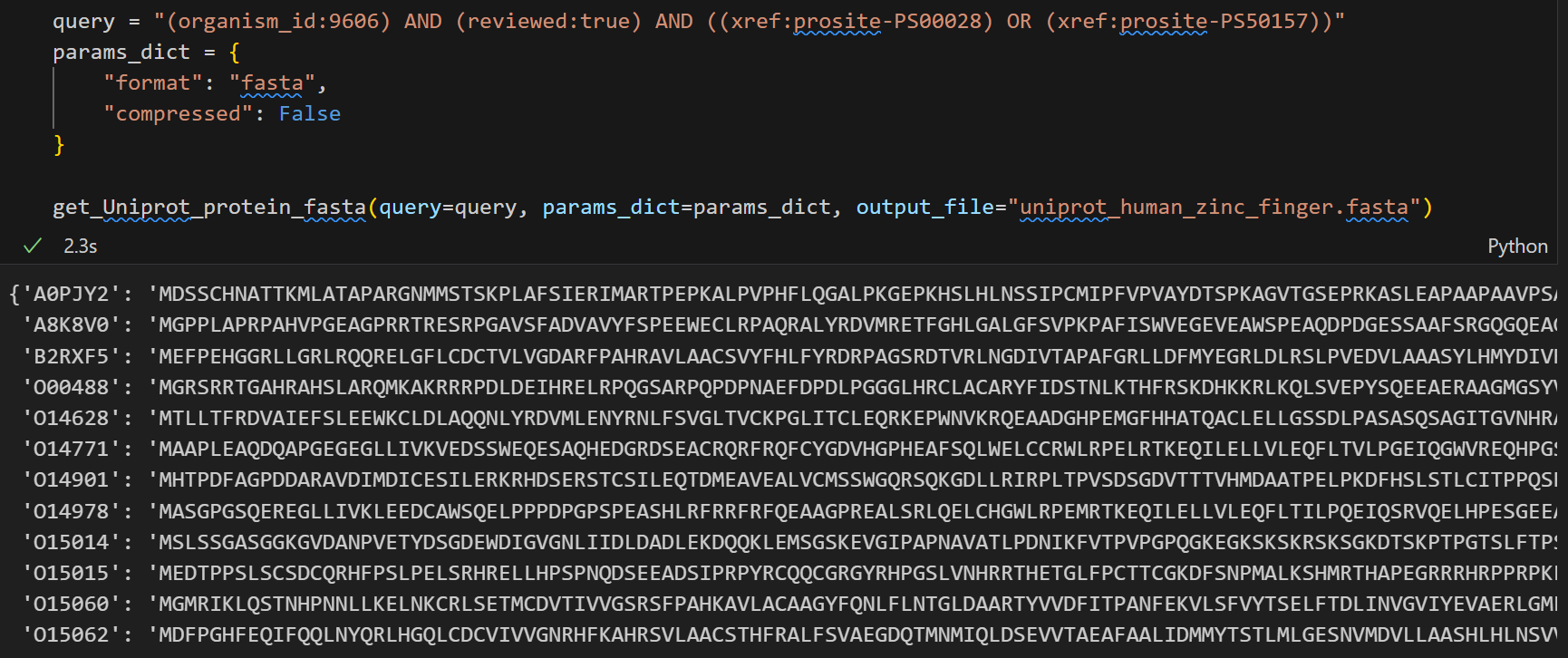
确实是791个,没有问题了
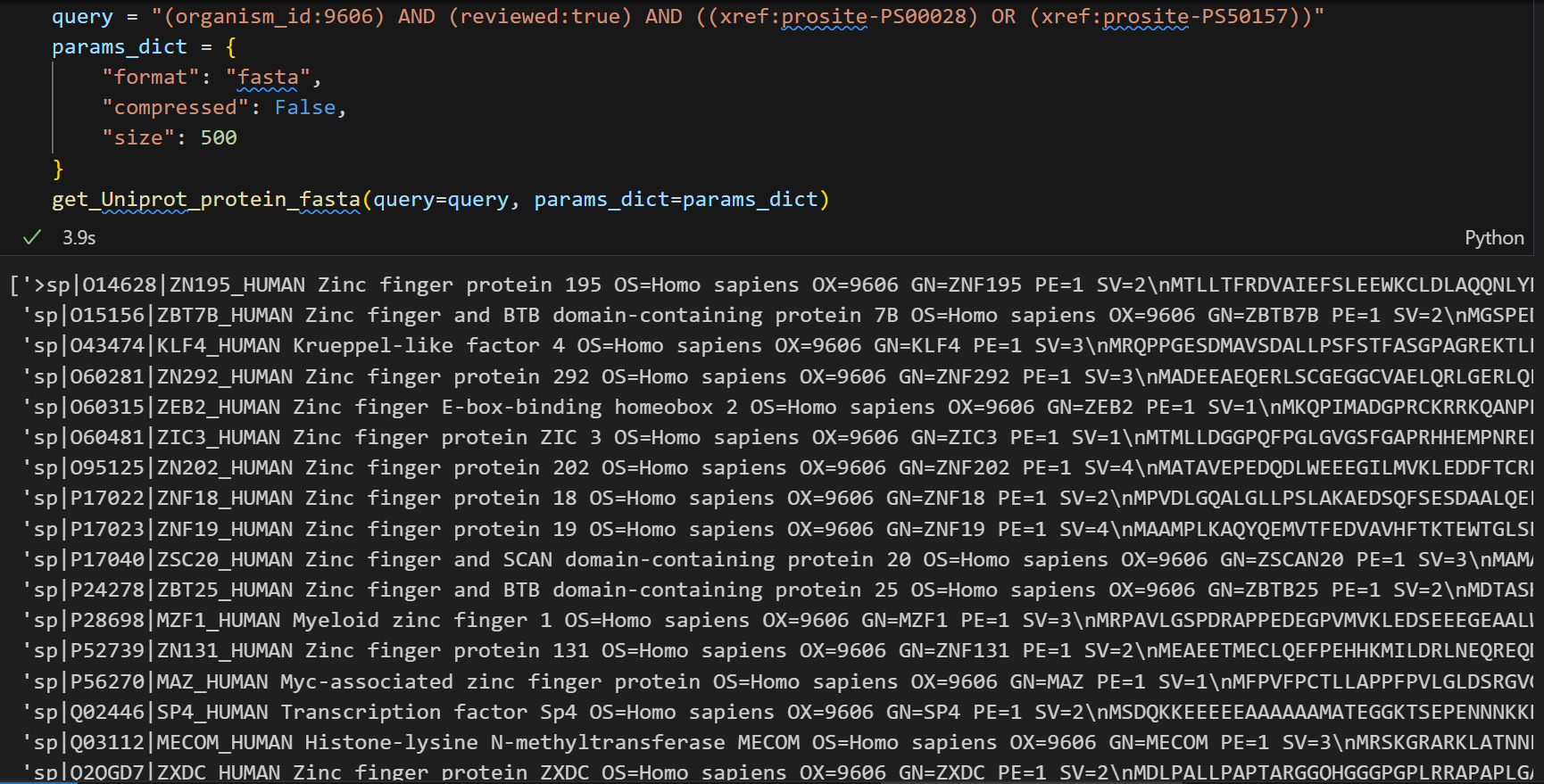
输出格式上有点问题,后面的几个条目头部都没有">"符合等,稍微再修改一下。
我们对于每一页page添加合并join之后的其实是一个大字符串,
类似效果如下:
requests.get("https://rest.uniprot.org/uniprotkb/search?query=P49711 and reviewed:true&format=fasta&compressed=false&size=500").text
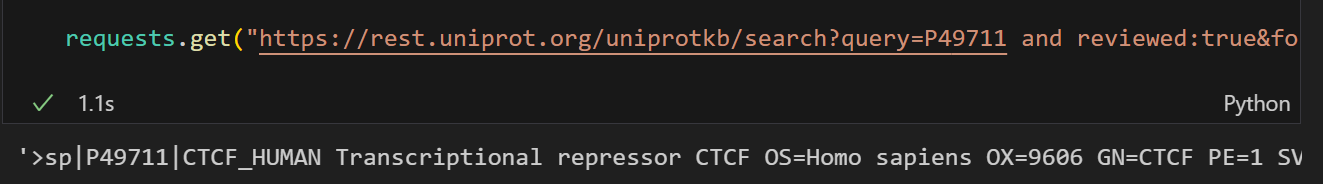
这个大字符串中,分割的依据就是按照换行符,
所以我们除了前面那种使用“\n>”(其实就是换行之后新开一个header,但是分割之后新的条目其实就没有>符号了),
就是使用正常的换行符来分割:
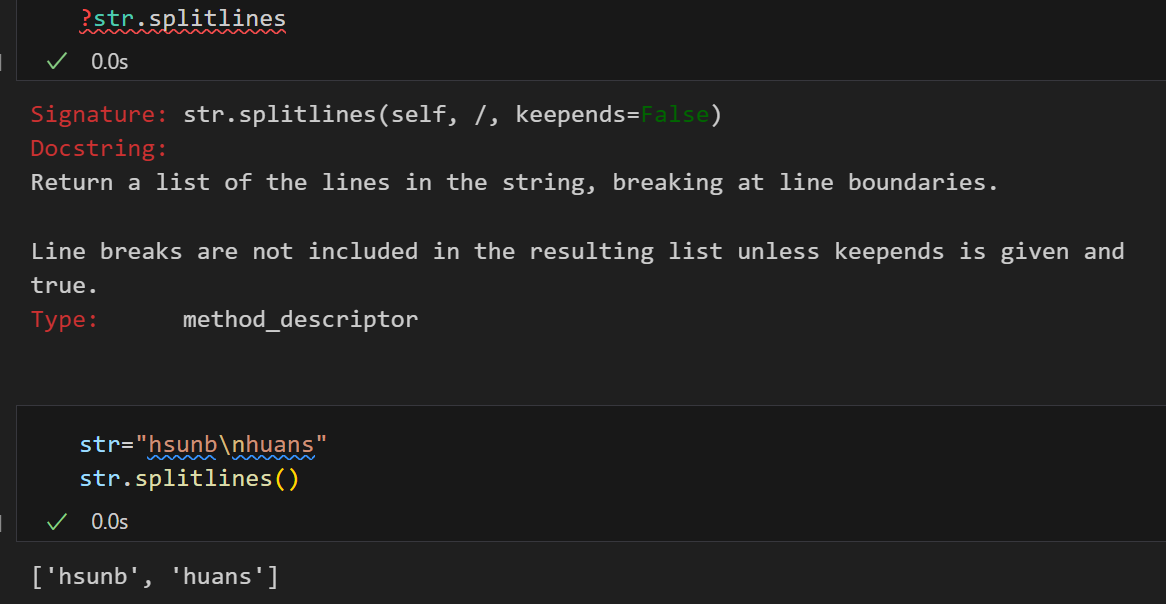
处理就是分割之后每一行按照fasta条目的header还是body进行处理:
import requests, time
def get_Uniprot_protein_fasta(query: str, params_dict: dict) -> list[str]:"""Description:根据给定的查询参数, 从Uniprot数据库中获取蛋白质的fasta序列, 支持分页请求;Args:query (str): 查询字符串, 用于指定搜索条件, 可以是单个序列数据,也可以是批量查询;params_dict (dict): 其他请求参数的字典, 一般包括以下键值对:format (str): 返回数据的格式, 默认为"fasta";compressed (bool): 是否请求压缩格式的数据, 默认为False;size (int): 每页返回的记录数/单次请求返回的序列数, 默认为500Returns:list[str]: 包含所有获取到的fasta序列的列表Notes:1, params_dict中的键值对会被添加到请求参数中, query参数会单独传递, 虽然参数单独传递, 但是最后都合并到URL中进行请求。2, query参数参考https://www.uniprot.org/help/query-fields, params_dict参数则比较固定, 参考Args部分说明;3, query参数一般建议带上物种, organism_id:9606; 以及是否reviewed:true等过滤条件"""base = "https://rest.uniprot.org/uniprotkb/search"# 将查询参数添加到params_dict中params_dict["query"] = query# 初始化请求url为基础url, 同时准备一个列表用于存储获取到的fasta序列片段pages, url = [], base# 分页请求循环,直到没有下一页while True:# 首次请求时使用params字典,后续请求的"下一页"url已包含所有必要参数,因此不再重复传入params参数# 请求超时设置为60秒r = requests.get(url, params=params_dict if url == base else None, timeout=60)# 检查是否请求成功,否则抛出异常r.raise_for_status()# 解析响应内容,将当前请求返回的fasta序列添加到列表中pages.append(r.text)# 解析下一页url# 先尝试获取 “next” 对应的子字典,若不存在则返回空字典;再从子字典中获取 “url”,若不存在则返回None(表示无下一页)next_url = r.links.get("next", {}).get("url")# 若无下一页,则跳出循环if not next_url:break# 更新url为下一页的url,继续请求# next_url已包含所有参数,无需重复传递url = next_url# 为避免过于频繁请求,稍作延时time.sleep(0.2)# 返回所有获取到的fasta序列片段, 拼接为一个完整的字符串all_fasta = "".join(pages).strip()if not all_fasta:return []# 存储单个条目、每一行line的缓冲区records, buffer = [], []for line in all_fasta.splitlines():if line.startswith(">"):# 说明新开了一个序列# 如果前面已经有序列了, 则合并并保存前面的序列(buffer,保存时补上换行符,其实就是将lines重新转回str)if buffer:records.append("\n".join(buffer))# 如果前面没有新的序列buffer = [line]else:buffer.append(line)# 处理最后一个序列if buffer:records.append("\n".join(buffer))return records
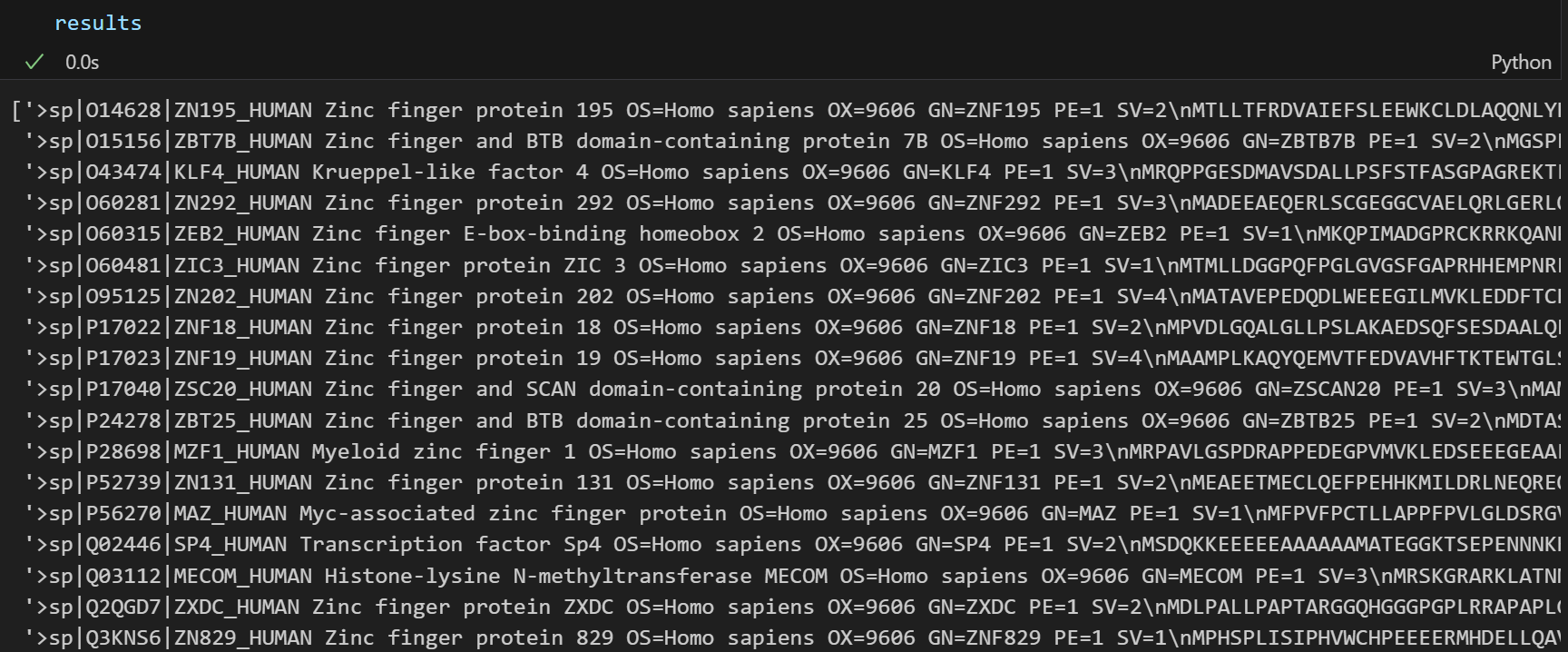
这下每一个结构都看着顺眼了。
当然,我们可以再处理一下,将结果返回为一个dict,ac:seq的字典。
就是对于结尾部分的代码再修改一下,如下:
import requests, time
def get_Uniprot_protein_fasta(query: str, params_dict: dict) -> list[str]:"""Description:根据给定的查询参数, 从Uniprot数据库中获取蛋白质的fasta序列, 支持分页请求;Args:query (str): 查询字符串, 用于指定搜索条件, 可以是单个序列数据,也可以是批量查询;params_dict (dict): 其他请求参数的字典, 一般包括以下键值对:format (str): 返回数据的格式, 默认为"fasta";compressed (bool): 是否请求压缩格式的数据, 默认为False;size (int): 每页返回的记录数/单次请求返回的序列数, 默认为500Returns:list[str]: 包含所有获取到的fasta序列的列表Notes:1, params_dict中的键值对会被添加到请求参数中, query参数会单独传递, 虽然参数单独传递, 但是最后都合并到URL中进行请求。2, query参数参考https://www.uniprot.org/help/query-fields, params_dict参数则比较固定, 参考Args部分说明;3, query参数一般建议带上物种, organism_id:9606; 以及是否reviewed:true等过滤条件"""base = "https://rest.uniprot.org/uniprotkb/search"# 将查询参数添加到params_dict中params_dict["query"] = query# 初始化请求url为基础url, 同时准备一个列表用于存储获取到的fasta序列片段pages, url = [], base# 分页请求循环,直到没有下一页while True:# 首次请求时使用params字典,后续请求的"下一页"url已包含所有必要参数,因此不再重复传入params参数# 请求超时设置为60秒r = requests.get(url, params=params_dict if url == base else None, timeout=60)# 检查是否请求成功,否则抛出异常r.raise_for_status()# 解析响应内容,将当前请求返回的fasta序列添加到列表中pages.append(r.text)# 解析下一页url# 先尝试获取 “next” 对应的子字典,若不存在则返回空字典;再从子字典中获取 “url”,若不存在则返回None(表示无下一页)next_url = r.links.get("next", {}).get("url")# 若无下一页,则跳出循环if not next_url:break# 更新url为下一页的url,继续请求# next_url已包含所有参数,无需重复传递url = next_url# 为避免过于频繁请求,稍作延时time.sleep(0.2)# 返回所有获取到的fasta序列片段, 拼接为一个完整的字符串all_fasta = "".join(pages).strip()if not all_fasta:return []# 存储单个条目、每一行line的缓冲区records, buffer = [], []for line in all_fasta.splitlines():if line.startswith(">"):# 说明新开了一个序列# 如果前面已经有序列了, 则合并并保存前面的序列(buffer,保存时补上换行符,其实就是将lines重新转回str)if buffer:records.append("\n".join(buffer))# 如果前面没有新的序列buffer = [line]else:buffer.append(line)# 处理最后一个序列if buffer:records.append("\n".join(buffer))# 我们可以再将每一个条目分别提取出来fasta_dict = {}for each_fasta in records:protein_id = each_fasta.split("|")[1]protein_seq = "".join(line.strip() for line in each_fasta.splitlines()[1:])fasta_dict[protein_id] = protein_seqreturn fasta_dict测试效果如下:
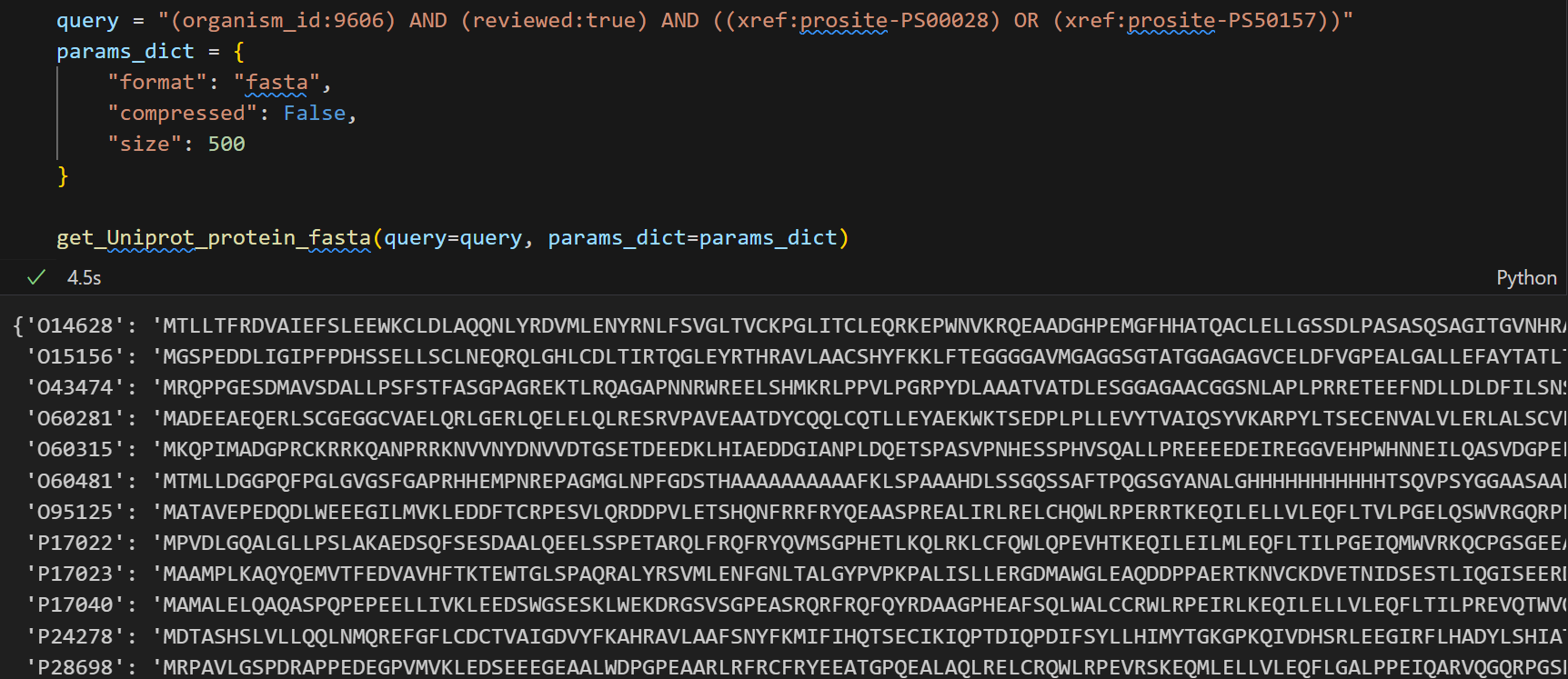 随便提取出哪一个的蛋白质序列出来:
随便提取出哪一个的蛋白质序列出来:
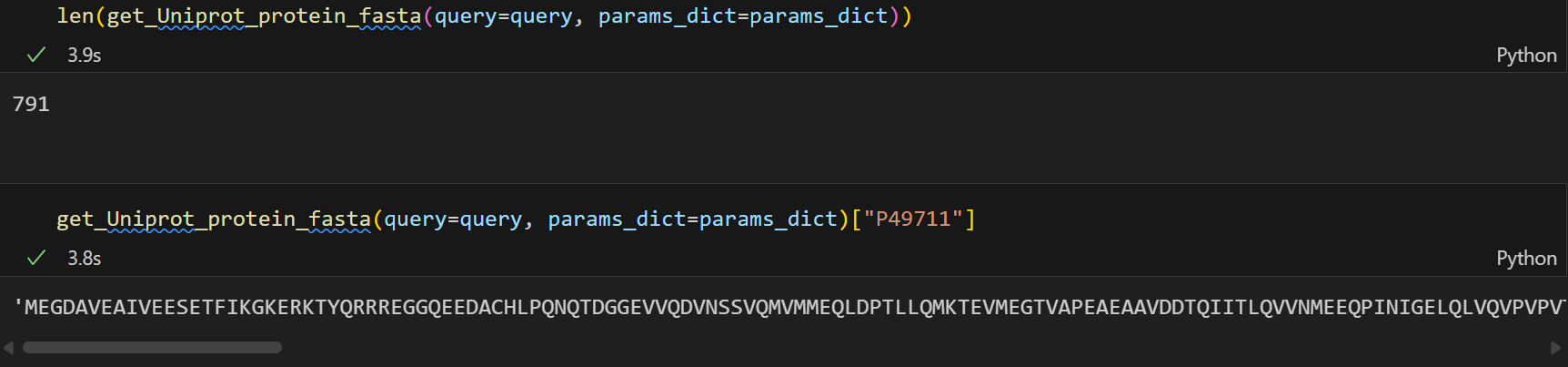
也可以将结果保存为fasta文件,我们再修改一下代码:
import requests, time
def get_Uniprot_protein_fasta(query: str, params_dict: dict, output_file: str) -> list[str]:"""Description:根据给定的查询参数, 从Uniprot数据库中获取蛋白质的fasta序列, 支持分页请求;Args:query (str): 查询字符串, 用于指定搜索条件, 可以是单个序列数据,也可以是批量查询;params_dict (dict): 其他请求参数的字典, 一般包括以下键值对:format (str): 返回数据的格式, 默认为"fasta";compressed (bool): 是否请求压缩格式的数据, 默认为False;size (int): 每页返回的记录数/单次请求返回的序列数, 默认为500output_file (str): 输出文件路径, 用于保存获取到的fasta序列;Returns:list[str]: 包含所有获取到的fasta序列的列表Notes:1, params_dict中的键值对会被添加到请求参数中, query参数会单独传递, 虽然参数单独传递, 但是最后都合并到URL中进行请求。2, query参数参考https://www.uniprot.org/help/query-fields, params_dict参数则比较固定, 参考Args部分说明;3, query参数一般建议带上物种, organism_id:9606; 以及是否reviewed:true等过滤条件"""base = "https://rest.uniprot.org/uniprotkb/search"# 将查询参数添加到params_dict中params_dict["query"] = query# 初始化请求url为基础url, 同时准备一个列表用于存储获取到的fasta序列片段pages, url = [], base# 分页请求循环,直到没有下一页while True:# 首次请求时使用params字典,后续请求的"下一页"url已包含所有必要参数,因此不再重复传入params参数# 请求超时设置为60秒r = requests.get(url, params=params_dict if url == base else None, timeout=60)# 检查是否请求成功,否则抛出异常r.raise_for_status()# 解析响应内容,将当前请求返回的fasta序列添加到列表中pages.append(r.text)# 解析下一页url# 先尝试获取 “next” 对应的子字典,若不存在则返回空字典;再从子字典中获取 “url”,若不存在则返回None(表示无下一页)next_url = r.links.get("next", {}).get("url")# 若无下一页,则跳出循环if not next_url:break# 更新url为下一页的url,继续请求# next_url已包含所有参数,无需重复传递url = next_url# 为避免过于频繁请求,稍作延时time.sleep(0.2)# 返回所有获取到的fasta序列片段, 拼接为一个完整的字符串all_fasta = "".join(pages).strip()if not all_fasta:return []# 存储单个条目、每一行line的缓冲区records, buffer = [], []for line in all_fasta.splitlines():if line.startswith(">"):# 说明新开了一个序列# 如果前面已经有序列了, 则合并并保存前面的序列(buffer,保存时补上换行符,其实就是将lines重新转回str)if buffer:records.append("\n".join(buffer))# 如果前面没有新的序列buffer = [line]else:buffer.append(line)# 处理最后一个序列if buffer:records.append("\n".join(buffer))# 我们可以再将每一个条目分别提取出来fasta_dict = {}for each_fasta in records:protein_id = each_fasta.split("|")[1]protein_seq = "".join(line.strip() for line in each_fasta.splitlines()[1:])fasta_dict[protein_id] = protein_seq# 将结果保存到一个指定的输出文件中with open(output_file,"w") as file:file.write(all_fasta)return fasta_dict
现在再次运行
query = "(organism_id:9606) AND (reviewed:true) AND ((xref:prosite-PS00028) OR (xref:prosite-PS50157))"
params_dict = {"format": "fasta","compressed": False,"size": 500
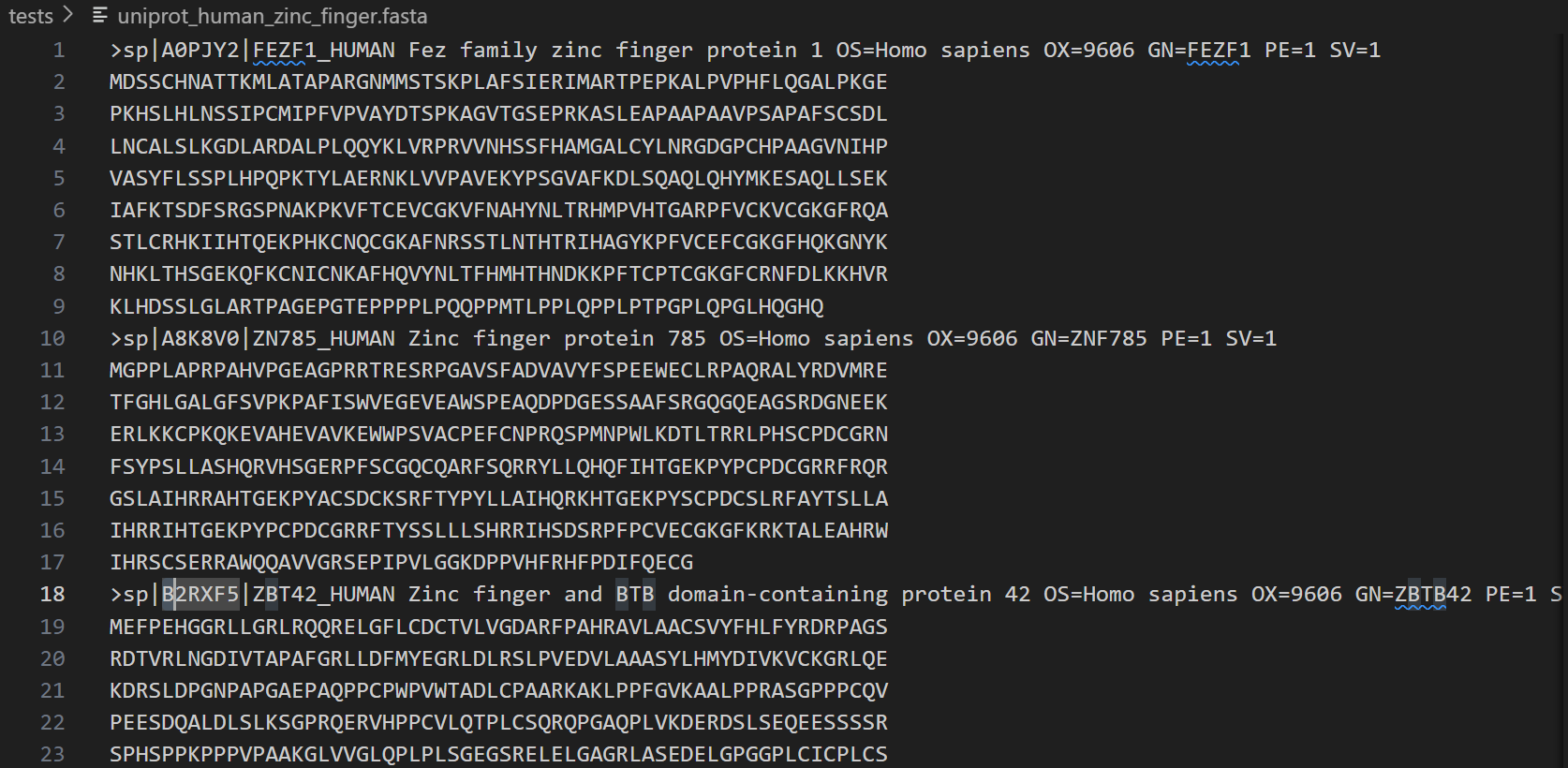
}get_Uniprot_protein_fasta(query=query, params_dict=params_dict, output_file="uniprot_human_zinc_finger.fasta")
下面这个就是我们输出的文件:
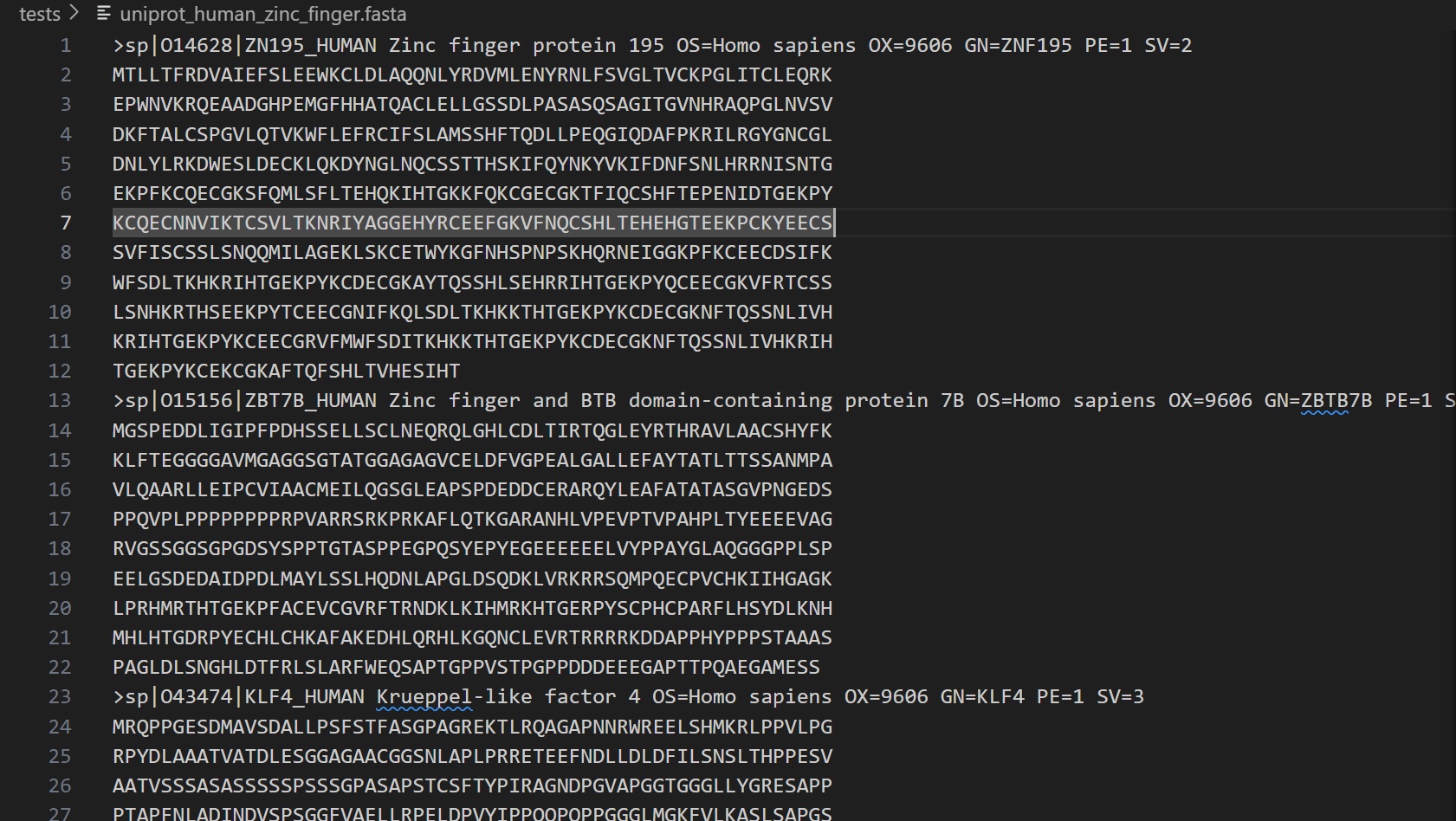
另外这里我需要重点强调一点的是:!!!
千万要注意,uniprot的数据库是会不断更新的,这也就意味着某一个accession序列号下的蛋白质,其序列是会不断变化的,
所以有时候保存出来的文件,有些细节上会不一样,这个时候,千万不要怀疑自己的代码有问题,在排除掉各种可能性之后,最不可能的可能就是肯定的。
这里举一个我自己的例子,我之前查询某一个某家族的蛋白质,同样在uniprot中,因为数目比较少,所以我当时并没有使用API来处理,直接在搜索框中依据query语法进行了手动搜索下载;
后来我用api用同样的关键词构建query,用脚本数据下载了一次;
两次数据之间差了4行:
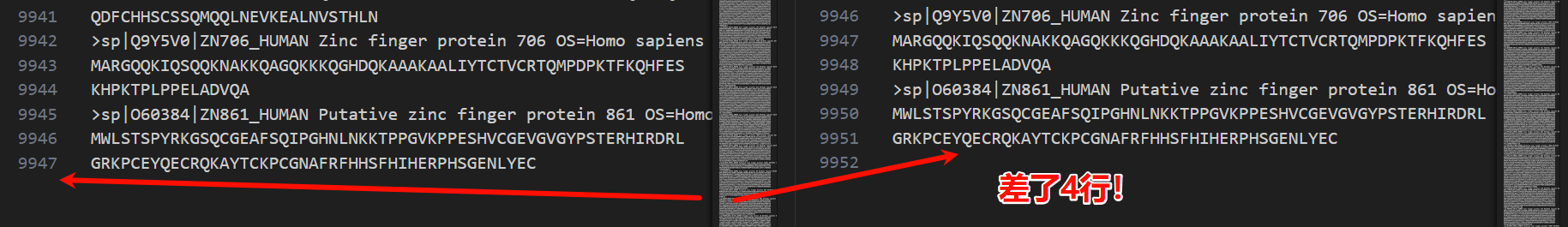
因为数据排序不一样,貌似脚本下载是按照页面顺序的,手动下载是按照id字母顺序的,所以不好直接比较;
我在排查了id,id的行数之后,发现有一个蛋白质的id有差异:
def compare_fasta_id_linecounts(file1: str, file2: str, include_blank: bool = False, show_top: int = 50):import sysdef count_seq_lines(path: str):counts = {}dups = set()cur_id, cur_cnt = None, 0def flush():nonlocal cur_id, cur_cntif cur_id is not None:if cur_id in counts:dups.add(cur_id)counts[cur_id] += cur_cntelse:counts[cur_id] = cur_cntcur_id, cur_cnt = None, 0with open(path, "r", encoding="utf-8", errors="ignore") as fh:for raw in fh:line = raw.rstrip("\r\n")if line.startswith(">"):parts = line.split("|", 2)flush()cur_id = parts[1] if len(parts) >= 2 else line[1:].split()[0]else:if include_blank or line != "":cur_cnt += 1flush()return counts, dupsc1, d1 = count_seq_lines(file1)c2, d2 = count_seq_lines(file2)if d1: print(f"[warn] {file1} 存在重复ID:{len(d1)}(已累计行数)", file=sys.stderr)if d2: print(f"[warn] {file2} 存在重复ID:{len(d2)}(已累计行数)", file=sys.stderr)all_ids = sorted(set(c1) | set(c2))diffs = []for i in all_ids:v1, v2 = c1.get(i, 0), c2.get(i, 0)if v1 != v2:diffs.append({"id": i, "file1_lines": v1, "file2_lines": v2, "diff": v1 - v2})print(f"IDs: file1={len(c1)}, file2={len(c2)}, union={len(all_ids)}, diffs={len(diffs)}")if show_top and diffs:print("id\tfile1_lines\tfile2_lines\tdiff(file1-file2)")for r in sorted(diffs, key=lambda x: abs(x["diff"]), reverse=True)[:show_top]:print(f"{r['id']}\t{r['file1_lines']}\t{r['file2_lines']}\t{r['diff']}")return diffs# 运行比较
diffs = compare_fasta_id_linecounts("/data2/file1.fasta","/data2/file2.fasta",include_blank=False, # 如需把空白行也计入行数,改为 Trueshow_top=100
)
len(diffs)

就是这个蛋白质,在旧数据中是8行,我用脚本下载之后变成了4行,所以少了那4行!
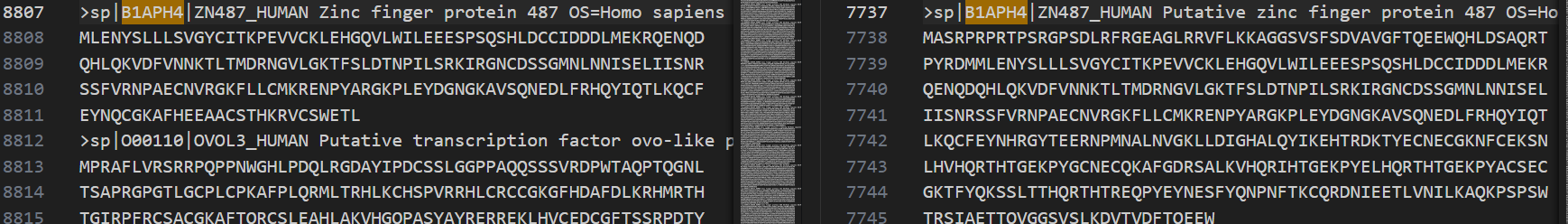
所以我就感觉很奇怪,为什么同一个accession id的数据会变化,
后来我到uniprot上一看,才发现在我两次获取数据之间,序列数据发生了更新!恍然大悟:
- 使用stream接口一次性流式返回所有条目
这个参考前面对于stream接口的说明,这个就比较简单了,因为不需要翻页
import requestsquery = "(organism_id:9606) AND (reviewed:true) AND ((xref:prosite-PS00028) OR (xref:prosite-PS50157))"
url = "https://rest.uniprot.org/uniprotkb/stream"
params = {"query": query, "format": "fasta", "compressed": "false"}r = requests.get(url, params=params, stream=True, timeout=60)
r.raise_for_status()
r.text
同样,我们可以写成一个脚本:
参考:https://github.com/MaybeBio/bioinfor_script_modules/blob/main/40_get_UniProt_protein_fasta.py
import requests, time
def get_Uniprot_protein_fasta(query: str, params_dict: dict, output_file: str) -> list[str]:"""Description:根据给定的查询参数, 从Uniprot数据库中获取蛋白质的fasta序列, 支持分页请求;Args:query (str): 查询字符串, 用于指定搜索条件, 可以是单个序列数据,也可以是批量查询;params_dict (dict): 其他请求参数的字典, 一般包括以下键值对:format (str): 返回数据的格式, 默认为"fasta";compressed (bool): 是否请求压缩格式的数据, 默认为False;size (int): 每页返回的记录数/单次请求返回的序列数, 默认为500output_file (str): 输出文件路径, 用于保存获取到的fasta序列;Returns:list[str]: 包含所有获取到的fasta序列的列表Notes:1, params_dict中的键值对会被添加到请求参数中, query参数会单独传递, 虽然参数单独传递, 但是最后都合并到URL中进行请求。2, query参数参考https://www.uniprot.org/help/query-fields, params_dict参数则比较固定, 参考Args部分说明;3, query参数一般建议带上物种, organism_id:9606; 以及是否reviewed:true等过滤条件"""base = "https://rest.uniprot.org/uniprotkb/stream"# 将查询参数添加到params_dict中params_dict["query"] = query# 使用流式端点进行请求r = requests.get(base, params=params_dict, stream=True, timeout=60)r.raise_for_status()if not r.text.strip():return []# 存储单个条目、每一行line的缓冲区records, buffer = [], []# 对于stream, 建议使用iter_lines方法逐行读取内容, 不使用splitlines方法for line in r.iter_lines(decode_unicode=True):if line.startswith(">"):# 说明新开了一个序列# 如果前面已经有序列了, 则合并并保存前面的序列(buffer,保存时补上换行符,其实就是将lines重新转回str)if buffer:records.append("\n".join(buffer))# 如果前面没有新的序列buffer = [line]else:buffer.append(line)# 处理最后一个序列if buffer:records.append("\n".join(buffer))# 我们可以再将每一个条目分别提取出来fasta_dict = {}for each_fasta in records:protein_id = each_fasta.split("|")[1]protein_seq = "".join(line.strip() for line in each_fasta.splitlines()[1:])fasta_dict[protein_id] = protein_seq# 将结果保存到一个指定的输出文件中with open(output_file,"w") as file:file.write(r.text)return fasta_dict为了更符合流式特点,以及我想在搜索大型序列时候提供输出文件便于输出,但是小型文件我不想输出,直接返回字典结果,
我又修改了一下脚本,
如果提供了输出文件,就输出,否则只返回字典结果:
def get_Uniprot_protein_fasta(query: str, params_dict: dict, output_file: Optional[str]) -> dict[str, str]:"""Description----------根据给定的查询参数, 从Uniprot数据库中获取蛋白质的fasta序列, 支持分页请求;Args----------query (str): 查询字符串, 用于指定搜索条件, 可以是单个序列数据,也可以是批量查询;params_dict (dict): 其他请求参数的字典, 一般包括以下键值对:- format (str): 返回数据的格式, 默认为 "fasta";- compressed (bool): 是否请求压缩格式的数据, 默认为 False;- size (int): 每页返回的记录数/单次请求返回的序列数, 默认为 500;output_file (str): 输出文件路径, 用于保存获取到的fasta序列, 如果为None则不保存文件, 建议查询大量序列时提供该参数, 如果只查询单个序列可以不提供;Returns----------Dict[str, str]: 包含所有获取到的fasta序列的字典, 键为蛋白质ID, 值为对应的fasta序列字符串;Notes----------- 1, params_dict中的键值对会被添加到请求参数中, query参数会单独传递, 虽然参数单独传递, 但是最后都合并到URL中进行请求。- 2, query参数参考https://www.uniprot.org/help/query-fields, params_dict参数则比较固定, 参考Args部分说明;- 3, query参数一般建议带上物种, organism_id:9606; 以及是否reviewed:true等过滤条件- 4, 该法取2, 使用stream端点进行请求, 避免分页复杂性, 详情参考https://github.com/MaybeBio/bioinfor_script_modules/blob/main/40_get_UniProt_protein_fasta.py中法12Example---------->>> query = "(organism_id:9606) AND (reviewed:true) AND ((xref:prosite-PS00028) OR (xref:prosite-PS50157))">>> params_dict = {... "format": "fasta",... "compressed": False... }>>> c2h2_zfp = get_Uniprot_protein_fasta(query=query, ... params_dict=params_dict, ... output_file="/data2/IDR_Pattern/data/raw/c2h2_zf_PROSITE.fasta")"""base = "https://rest.uniprot.org/uniprotkb/stream"# 将查询参数添加到params_dict中params_dict["query"] = query# 使用流式端点进行请求r = requests.get(base, params=params_dict, stream=True, timeout=60)r.raise_for_status()# 存储单个条目、每一行line的缓冲区records, buffer = [], []# 对于stream, 建议使用iter_lines方法逐行读取内容, 不使用splitlines方法for line in r.iter_lines(decode_unicode=True):if line is None:continueif line.startswith(">"):# 说明新开了一个序列# 如果前面已经有序列了, 则合并并保存前面的序列(buffer,保存时补上换行符,其实就是将lines重新转回str)if buffer:records.append("\n".join(buffer))# 如果前面没有新的序列buffer = [line]else:buffer.append(line)# 处理最后一个序列if buffer:records.append("\n".join(buffer))# 我们可以再将每一个条目分别提取出来fasta_dict = {}for each_fasta in records:protein_id = each_fasta.split("|")[1]protein_seq = "".join(line.strip() for line in each_fasta.splitlines()[1:])fasta_dict[protein_id] = protein_seq# 将结果保存到一个指定的输出文件中(可选)if output_file:with open(output_file,"w") as file:# 最好不用r.text, 与流式请求不一致file.write("\n".join(records) + "\n")return fasta_dict
query = "(organism_id:9606) AND (reviewed:true) AND ((xref:prosite-PS00028) OR (xref:prosite-PS50157))"
params_dict = {"format": "fasta","compressed": False
}get_Uniprot_protein_fasta(query=query, params_dict=params_dict, output_file="uniprot_human_zinc_finger.fasta")
获取的fasta序列文件也是不错的:
(2)有了蛋白质id,想获取对应蛋白质的某些属性数据
比如说我手头上只有某个蛋白质的uniprot ID,然后我想通过这个ID获取各种信息,
比如说我们这篇博客的重点就是要获取其结构域数据。
我们还是以CTCF为例子,我知道它是P49711的id,
那么依据前面一(检索单个条目),
https://rest.uniprot.org/uniprotkb/P49711.fasta
就是我们的目标API,
但是如果我只是简单获取这个响应的内容的话,
实际的输出是:


按照前面的说法,1️⃣是Accession Number(ac),2️⃣是entry_name,3️⃣是sequence,
我们可以发现3️⃣就在第一个换行符后面,
ac在第1和第2个竖线符号中间,
我们可以使用正则表达式处理,假设我们想要返回(ac,entry,sequence)的3元元组数据。
最好的效果就是使用splitlines(),和文本格式展示效果一致;
或者用iter_lines见前
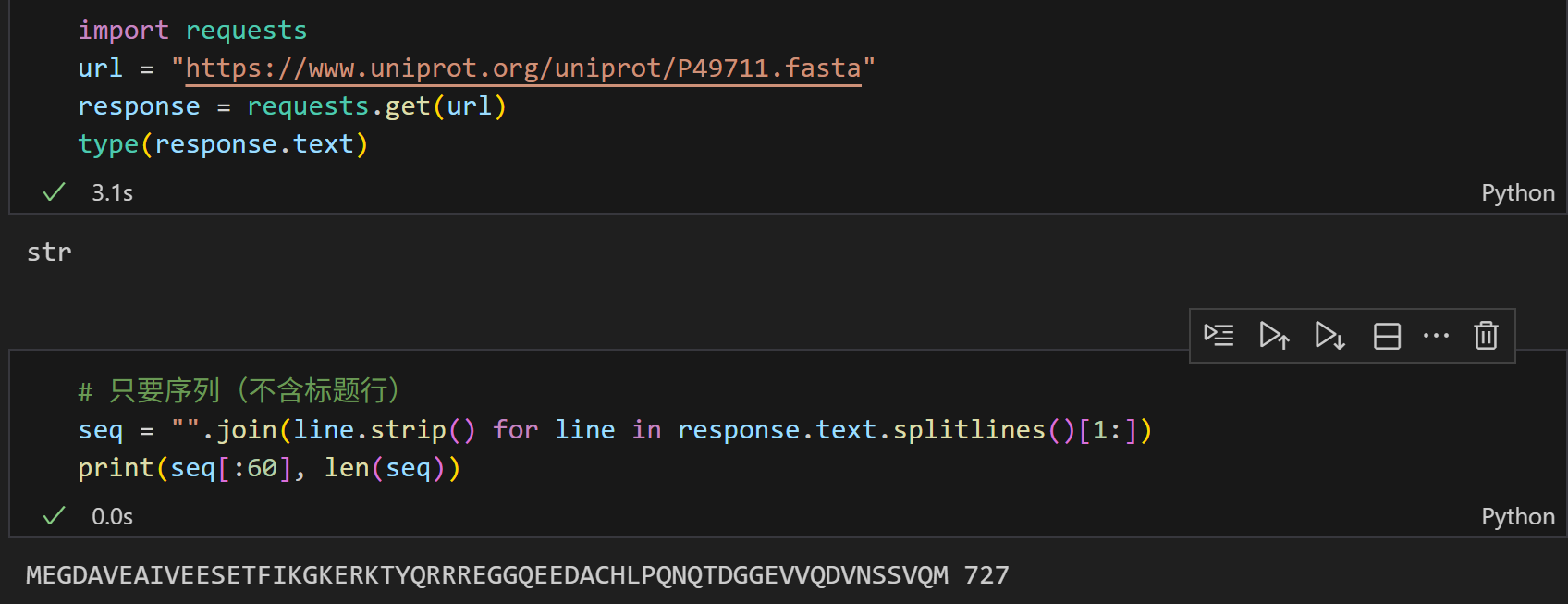
这样,一个非常简单的脚本就写好了:
import requestsdef fetch_Uniprot_protein_fasta(uniprot_id):"""输入uniprot id,获取其fasta序列数据"""# uniprot API base URLbase_url = "https://rest.uniprot.org/uniprotkb"# 构建query URLquery_url = f"{base_url}/{uniprot_id}.fasta"print(f"Downloading {uniprot_id}...")# 发送GET请求response = requests.get(query_url)if response.status_code == 200:# 请求成功,解析响应内容fasta_str = response.text# 依次解析ac,entry_name,sequenceid = fasta_str.split("|")[1]entry_name = fasta_str.split("|")[2].split(" ")[0]sequence = "".join(line.strip() for line in response.text.splitlines()[1:])return (uniprot_id,id,entry_name,sequence)else:# 请求失败,返回错误信息print(f"Failed to fetch fasta for {uniprot_id}. Status code: {response.status_code}")
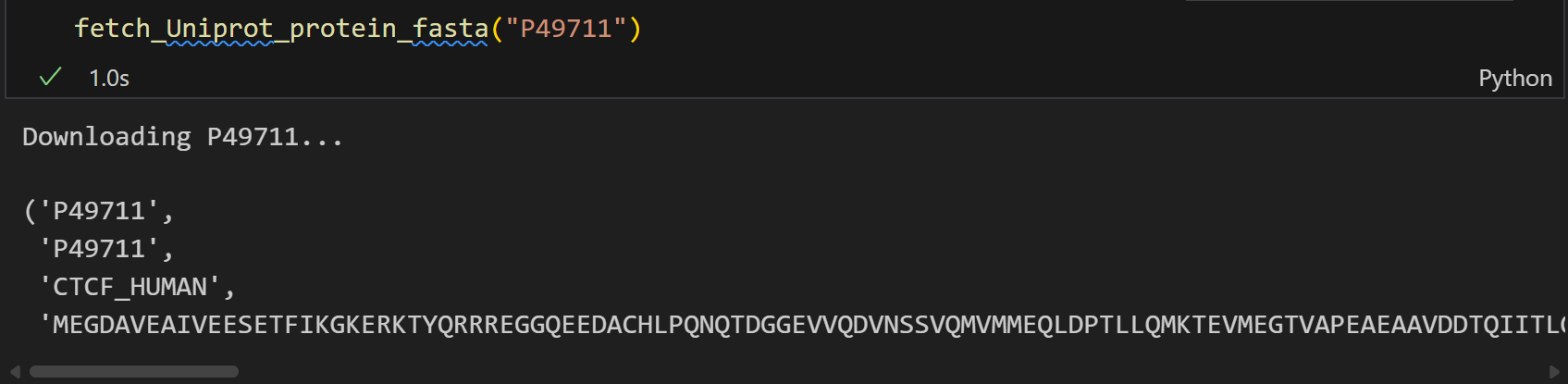
当然,这里元组前面的两个id我只是为了验证获取的数据是否一致,实际过程中都可以去掉。
当然,上面这些都不是重点,重点是我们需要获取这个蛋白质id的某些性质数据,
比如说我们这篇博客的重点,就是要获取结构域数据:
简单来说,我们就是要抓取下面的数据
我个人习惯于指定服务器返回数据格式为JSON,便于后续解析,从中获取结构域数据。
我们还是以CTCF为例,
import requests,time
url = "https://rest.uniprot.org/uniprotkb/P49711"
headers = {"Accept": "application/json"}response = requests.get(url, headers=headers)
response.json()
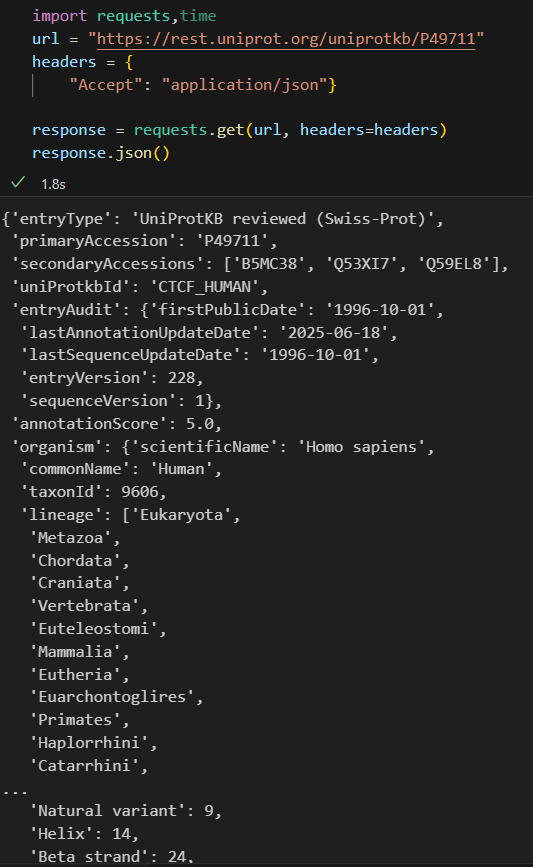
(在请求头中指定服务器返回json格式数据,返回的field等数据会比较多、更加丰富)。
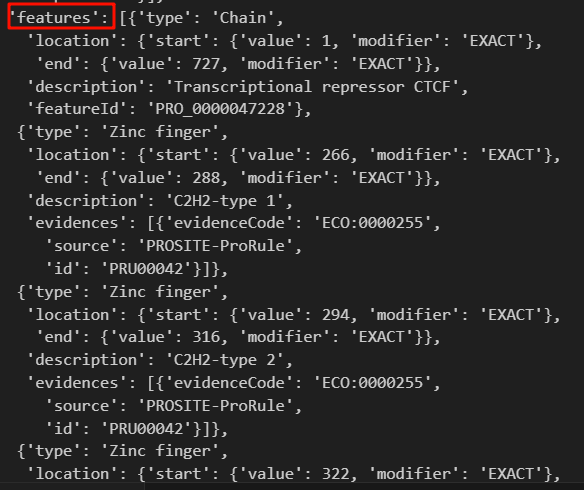
可以看到,features是我们想要的field,
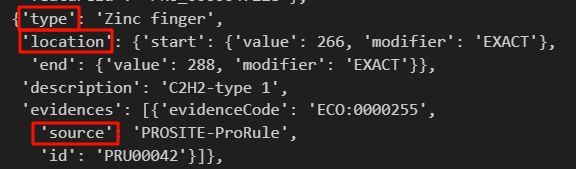
我还是以CTCF为例子,比如说这个蛋白,其中一个结构域:
- 首先type我们是需要的,我们需要知道这个是什么结构域,是不是结构域;
- 其次是location,这个是我们本次所需要的数据,基本上type id + location就是bed格式3要素
- source:来源于哪一个数据库,也就是是哪里注释的,需要标明,也是我们需要的
- 还有一个条目,是交叉参考crossreference:我们主要是关注其中的PDB、
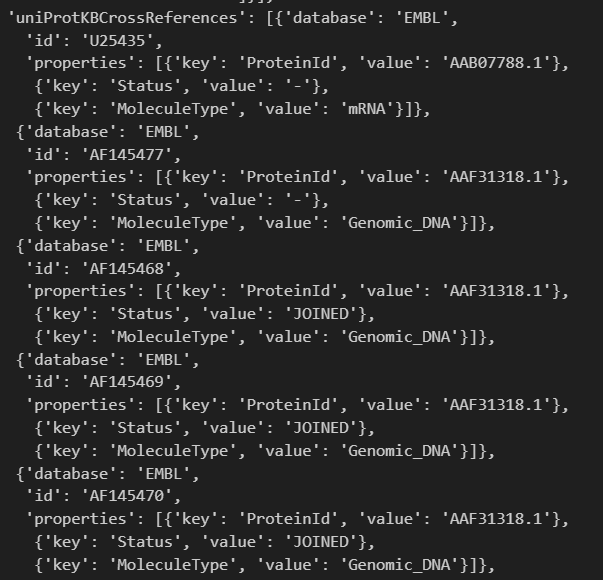
PDB给出了残基范围数据,我们可以获取response的时候顺便获取全场,然后在这里做一个判断,判断哪一个PDB的entry是包含全长的结构数据。
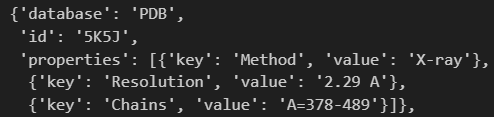
还有功能注释:
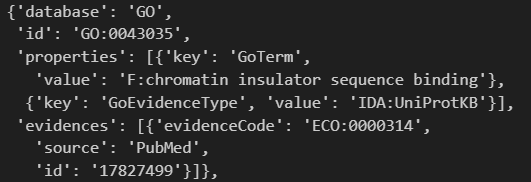
另外这里其实有其他的交叉参考数据库的,但是却没有显示其注释的区域信息:
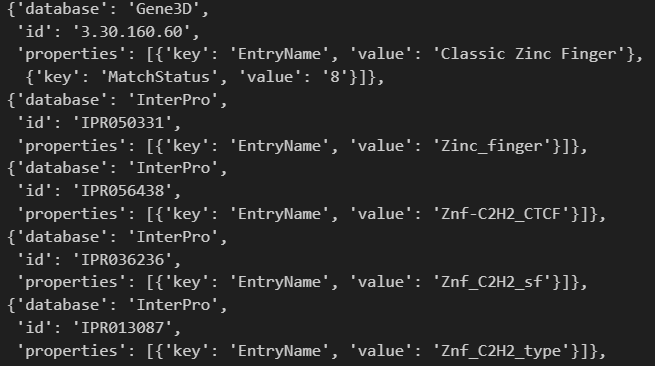
其实都只是hit数值,并不是区域坐标数据
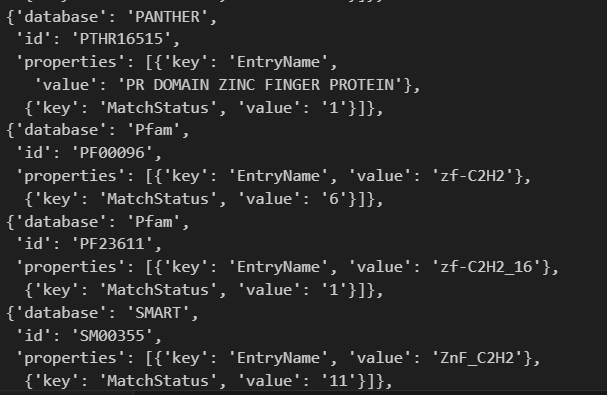
一个是8hit,一个是11hit,确实就像web页面显示的那样,因为web页面其实也没有展示具体的坐标数据,只能到具体的数据库中使用api,或者是用InterPro集成的api,参考下文。
至于其他的注释数据:
我放一个其他区域的注释,因为数据来源很多,所以我们要对上面这几个元素进行限制:
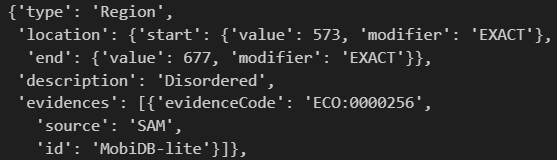
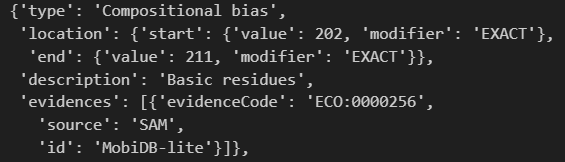
这里的这个修饰,其实就是PTM翻译后修饰了,也可以获取,不过我们有其他的数据库以及预测工具的接口,可以专门用于处理并获取这个数据。
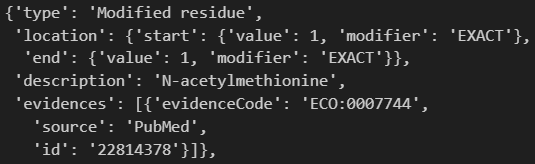
后面这个其实是二级结构了,这个其实可以用于一些结构相关的深度学习的数据收集,不过这个不是我们此次博客的重点。
而且这里还给出了PDB的序列号,其实可以用于后续抓取结构数据(空间坐标)。
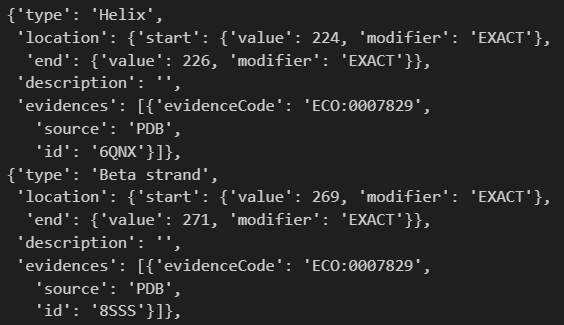
总之,回到我们的问题,其实这里的features字段就是各种区域注释的合集,
包括结构域、功能注释、PTM以及二级结构数据等等。
我们的目的就是在这个数据中进行挖掘:
import requests,time
url = "https://rest.uniprot.org/uniprotkb/P49711"
headers = {"Accept": "application/json"}response = requests.get(url, headers=headers)
data = response.json()
data['features']
然后我们的目的就是依据type类型将我们所需要的区域数据提取出来,
我们可以直接查看一下具体有哪些type类型的数据,比如说这里的CTCF
import requests,time
url = "https://rest.uniprot.org/uniprotkb/P49711"
headers = {"Accept": "application/json"}response = requests.get(url, headers=headers)
data = response.json()
for feature in data['features']:print(feature["type"])
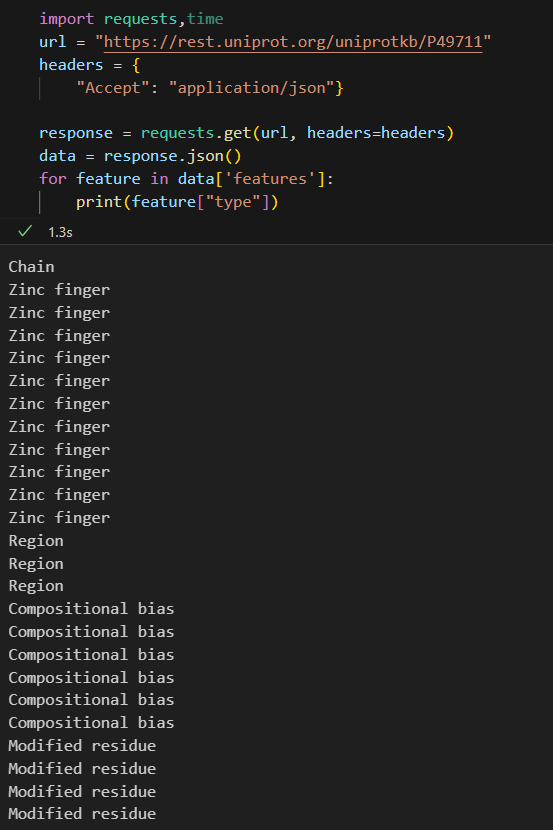
我们可以看到
Zinc finger是我们这里所需要的结构域数据Region其实是IDR, 而且注释也是MobiDB-lite算法注释的, 注意这个其实是古早注释了,
虽然数据库在不断更新, 但是benchmark上其实是比不过现在的深度学习方法了Modified residue其实是PTM, 也就是翻译后修饰
但是一个条目数据太少了,我们可以获取整个群体query的返回数据,查看其中的具体数据:
我们可以发现,其实results中就是每一个条目entry数据的集合,
import requests,time
# 使用stream接口获取数据
url = "https://rest.uniprot.org/uniprotkb/stream"query = "(organism_id:9606) AND (reviewed:true) AND ((xref:prosite-PS00028) OR (xref:prosite-PS50157))"headers = {"Accept": "application/json"}params = {}
params['query'] = queryresponse = requests.get(url, headers=headers, stream=True, params=params)data = response.json() len(data["results"])
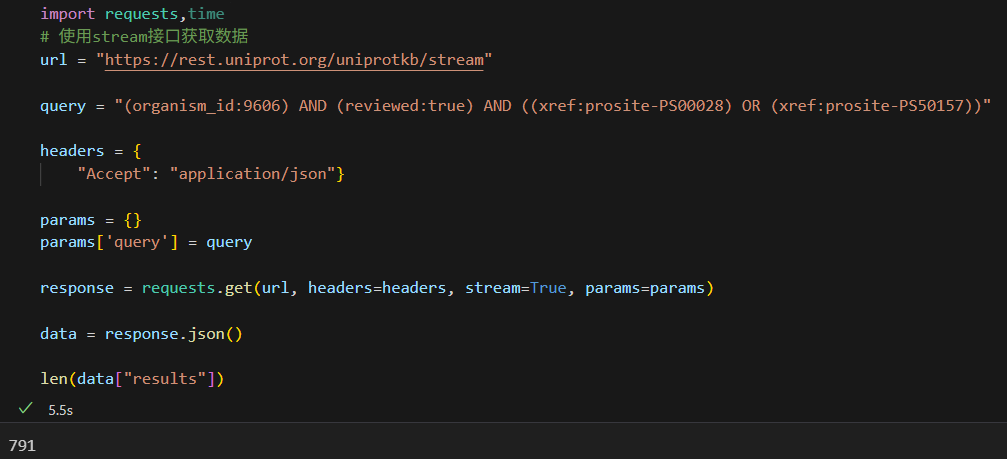
果然,其实results中的每一个迭代对象,就是一个单个蛋白质条目entry的网页,
可以从中获取序列号,我们查看一下:
for entry in data["results"]:print(entry["primaryAccession"])
既然每一个entry都是前面类似CTCF一样的结果,我们就可以在entry中展开查看features了:
for entry in data["results"]:print(entry["features"])
下面数据输出的每一行,都是前面类似于CTCF的结果
我们收集一下这个query所返回的群体性数据,其feature类型到底有哪些:
我们用集合来看,便于去重
fea_type_list = []
for entry in data["results"]:for feature in entry["features"]:fea_type_list.append(feature["type"])
set(fea_type_list)
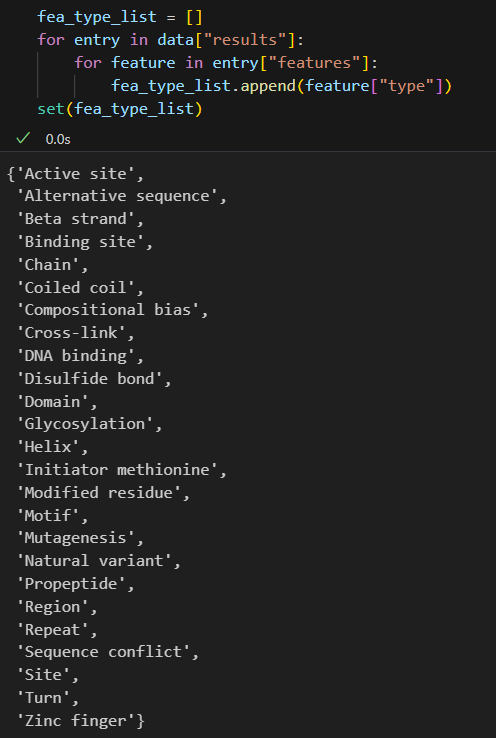
可以看到,虽然我们返回的条目数据很多,但是实际上归并之后注释的feature类型总共就那么十几个!
这些条目,我们需要结合官方文档+实际参考数据,来分析一下到底哪些是我们需要的,哪些不是:
链接参考:https://www.uniprot.org/help/family_and_domains_section

其实按照下面的概念:
domain、repeat、region、motif等都是我们所关注的,
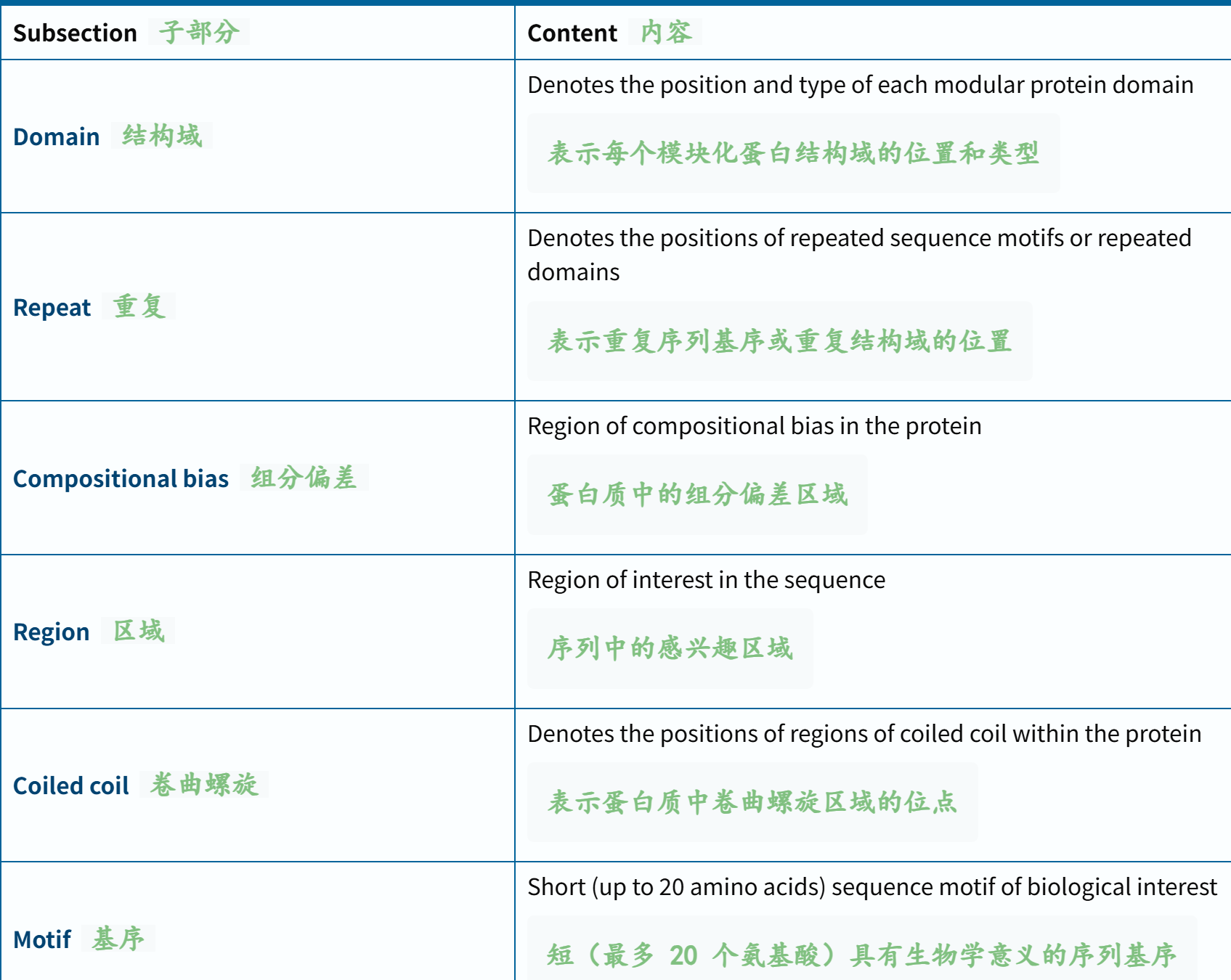

另外参考:https://www.uniprot.org/help/domain

我们可以看到,结构域注释管理,其实基本上都是InterPro数据库注释(InterPro数据联盟中的子数据库,比如说PROSITE,这个我们前面的CTCF就是例子;Pfam,纯粹就是MSA+HMM,其实不是很准确,但是以序列为输入倒是可行;SMART,比较全面,具体我会在下一个大节进行讨论)。

这里给的几个蛋白质示例都挺简洁的:
https://www.uniprot.org/uniprotkb/Q52107/entry#family_and_domains
domain也是PROSITE注释的
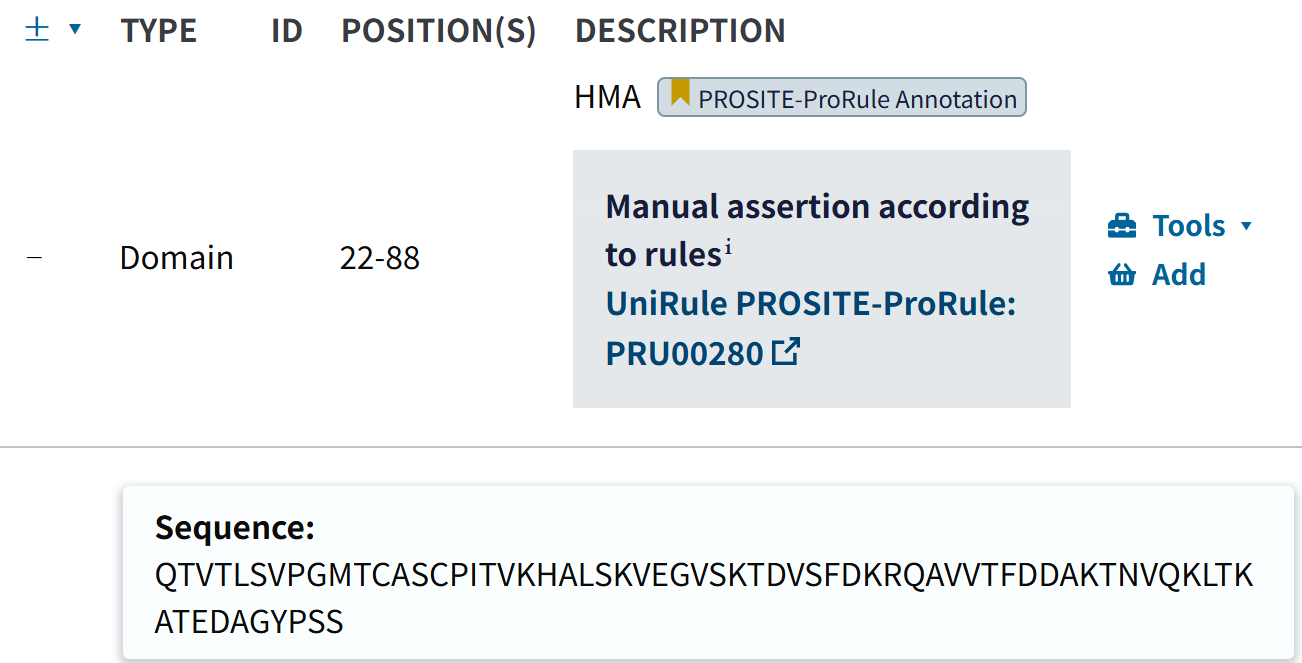
https://www.uniprot.org/uniprotkb/Q8TDF5/entry#family_and_domains
这个其实也没什么好说的,domain是PROSITE注释的,
但是下面的这个motif就很奇怪了,奇怪在于没有注释,不知道是哪里来的?
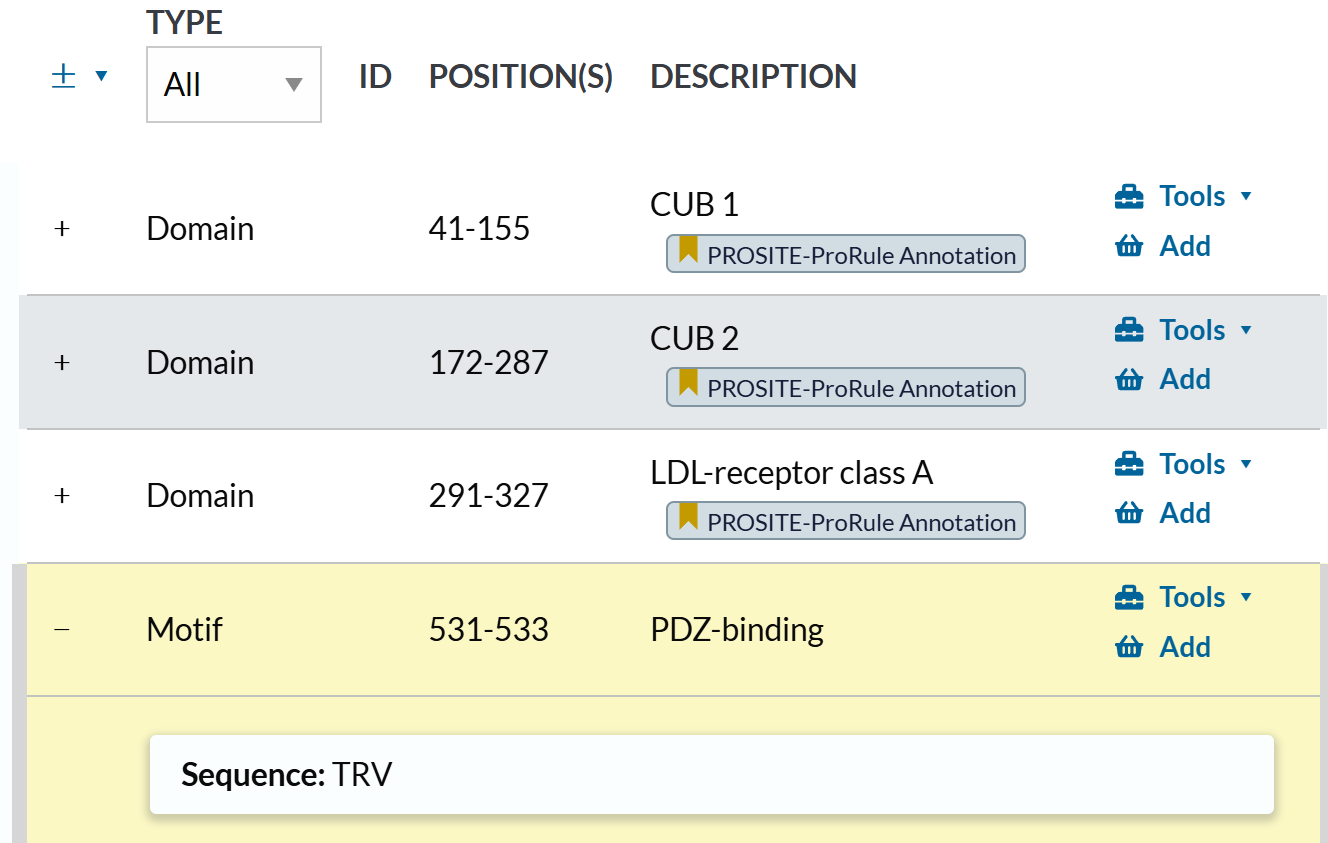
害得我还得看文献翻一翻这玩意是什么,
文献参考链接:https://pmc.ncbi.nlm.nih.gov/articles/PMC2652390/

其实结合坐标和图示,我们可以发现这个motif就在C端:
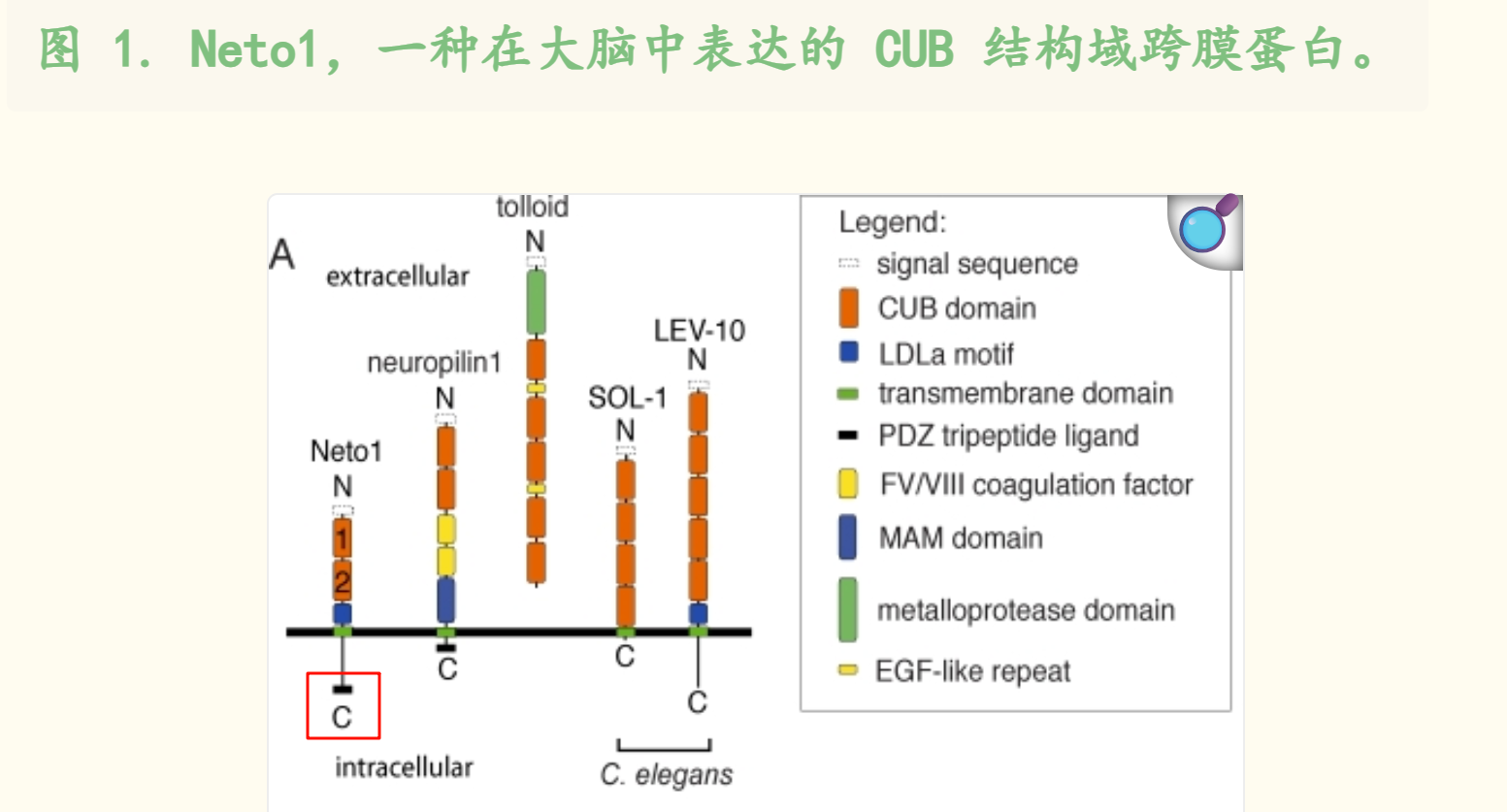
总之,motif这个数据虽然不是结构域,但是或多或少也是我们所需要的。
https://www.uniprot.org/uniprotkb/Q05193/entry#family_and_domains
这个条目里的domain,没什么好说的,也还是prosite注释的;
至于Region,出现了除disorder之外的,其他的类型,就是这里的motif类
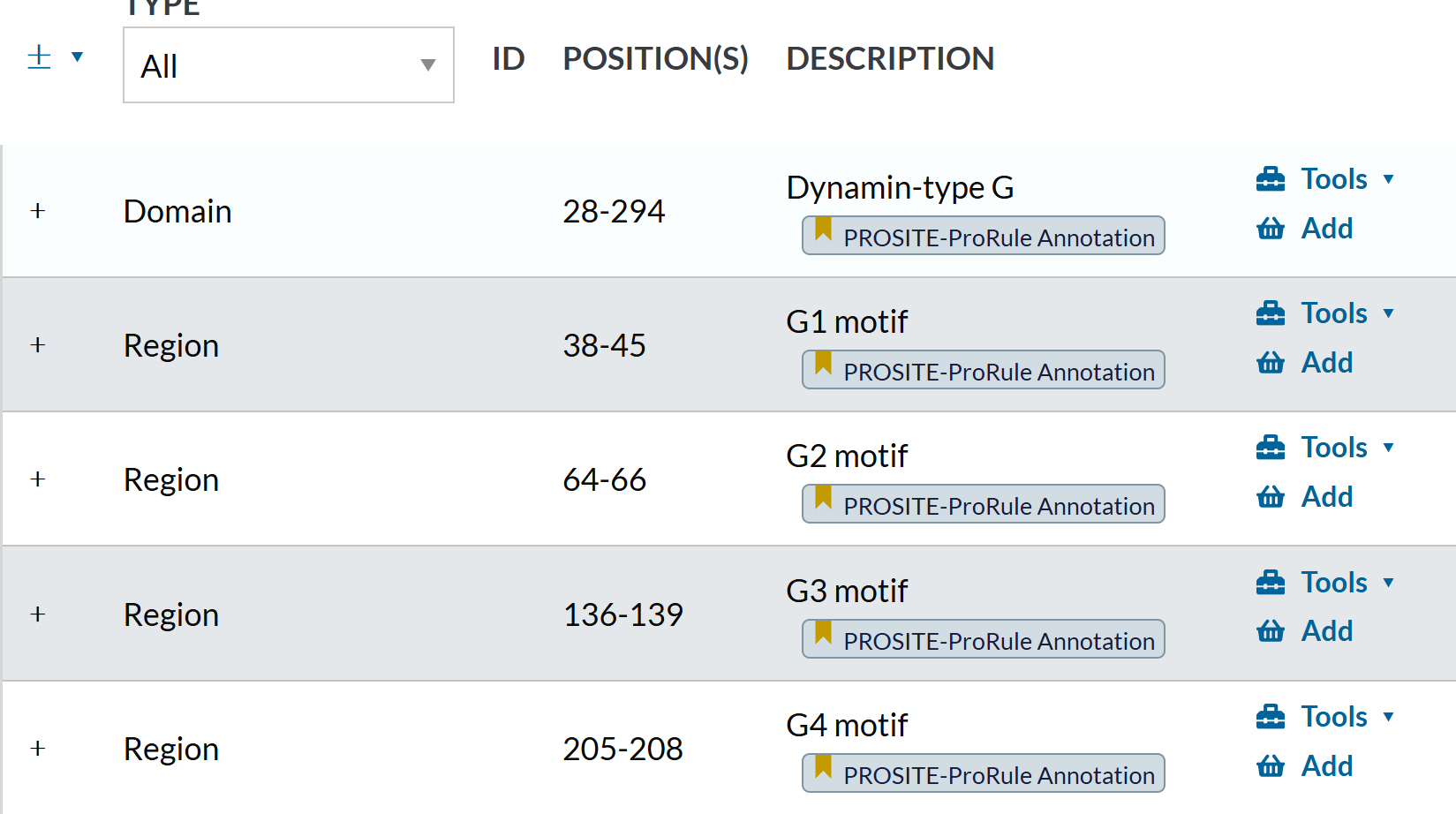
后面有disorder的region:

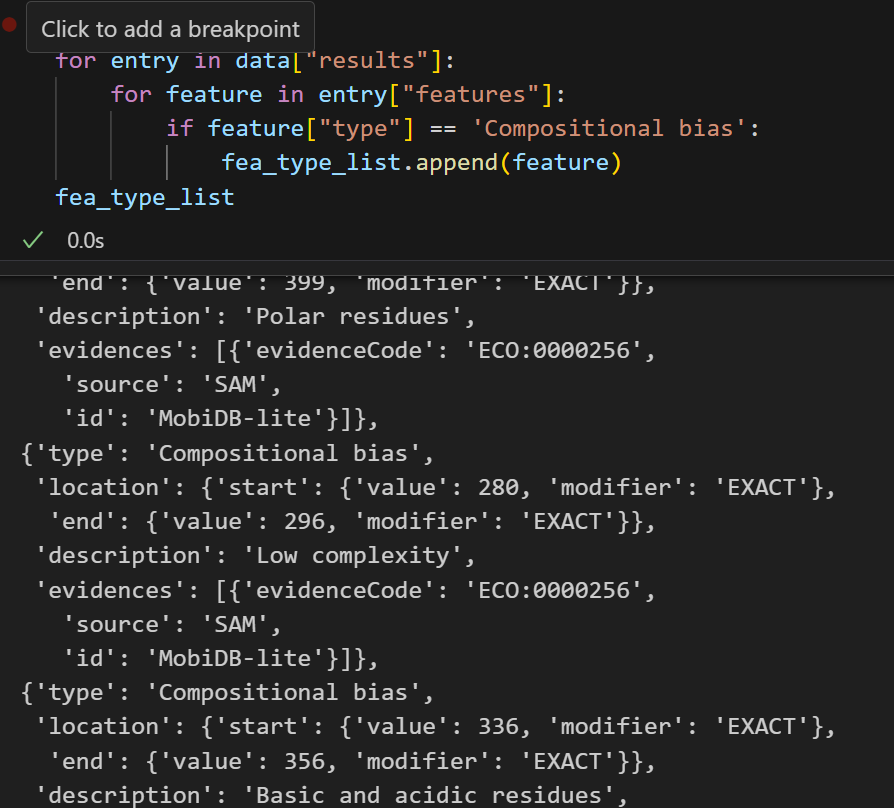
至于Compositional bias:
其实就是序列组成有偏差,也就是bias的区域,这个其实可以靠单纯的序列分析进行判断,而且是古早纯生信时代就有的分析内容,近几年已经很少看到有纯分析序列的了,大概是内容太少不足以支撑发一篇,或者是发在bioinformatics上。
总之,数据库里的注释也是依据序列分析自动注释。
其实从概念上来讲,IDR/LCR以及序列局部bias区域,都可以放到同一类中,比如说是IDR
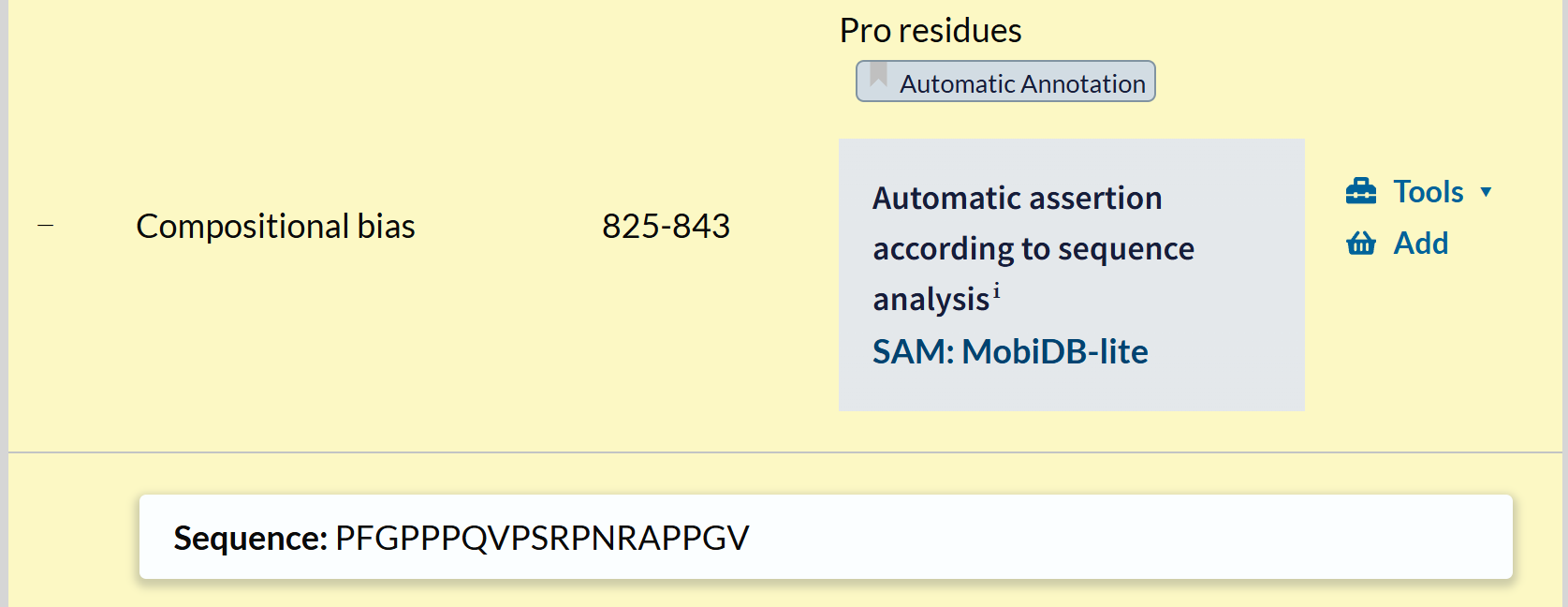
所以我们其实可以发现:
这个bias区域是在disorder region内

https://www.uniprot.org/uniprotkb/Q8BY02/entry#family_and_domains
这个条目下的数据:
Region还多了个单纯从序列分析,也就是相似性搜索中获取的注释
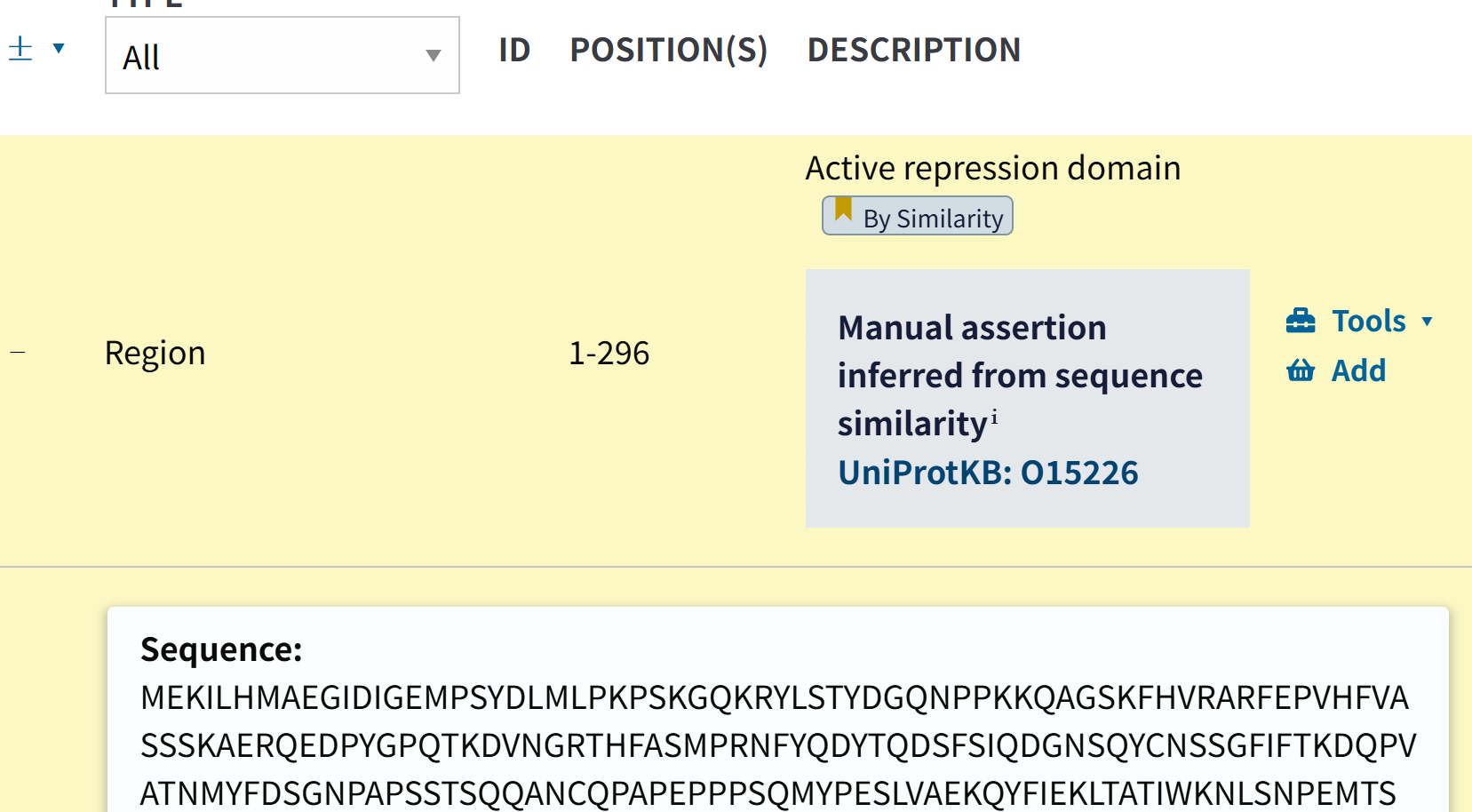
motif也有其他的类型,比如说是核定位信号,
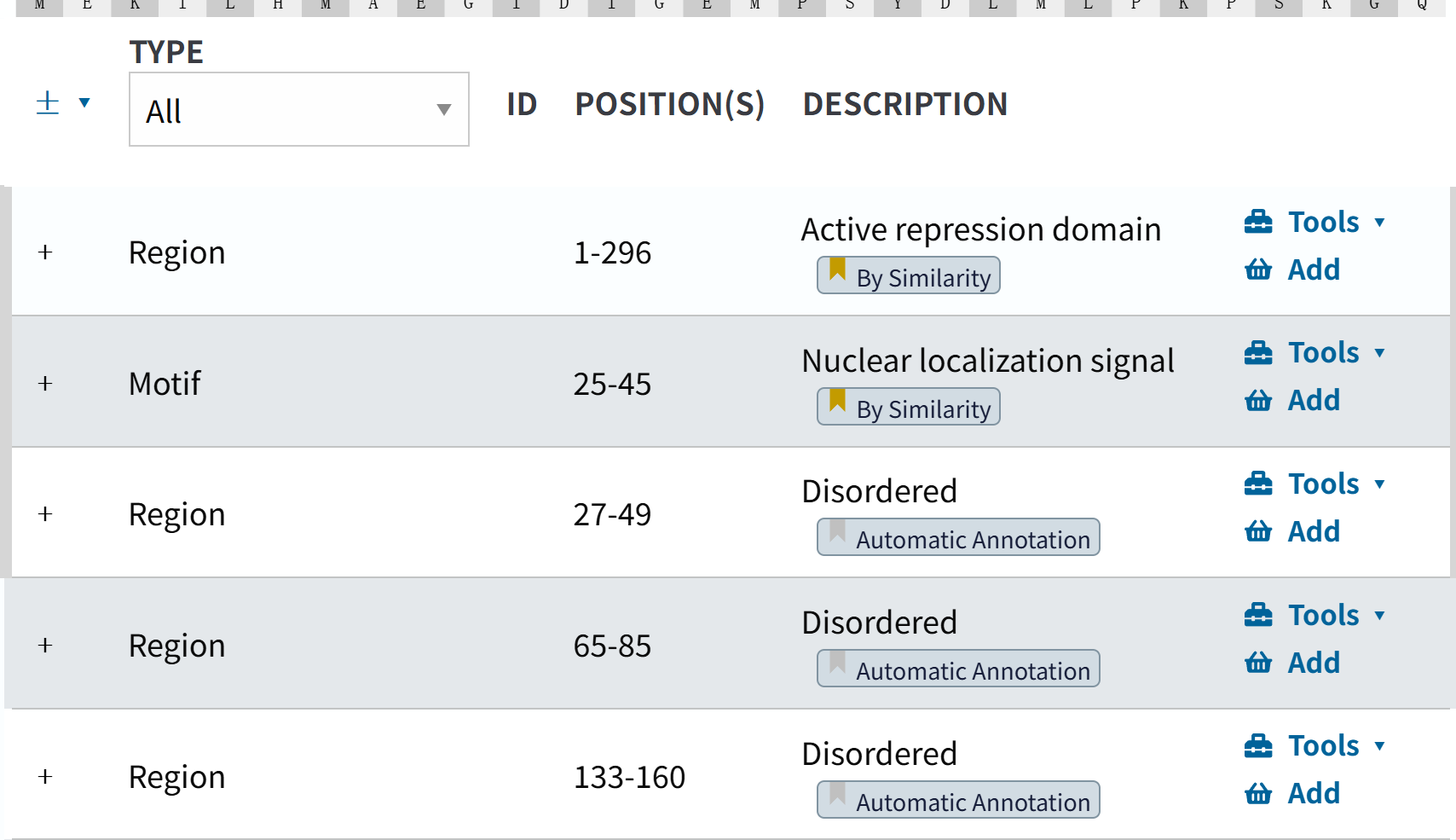
这个也可以囊括在我们的结构域数据研究中。
其余的区域注释,大部分要么是PROSITE注释,要么是自动注释。
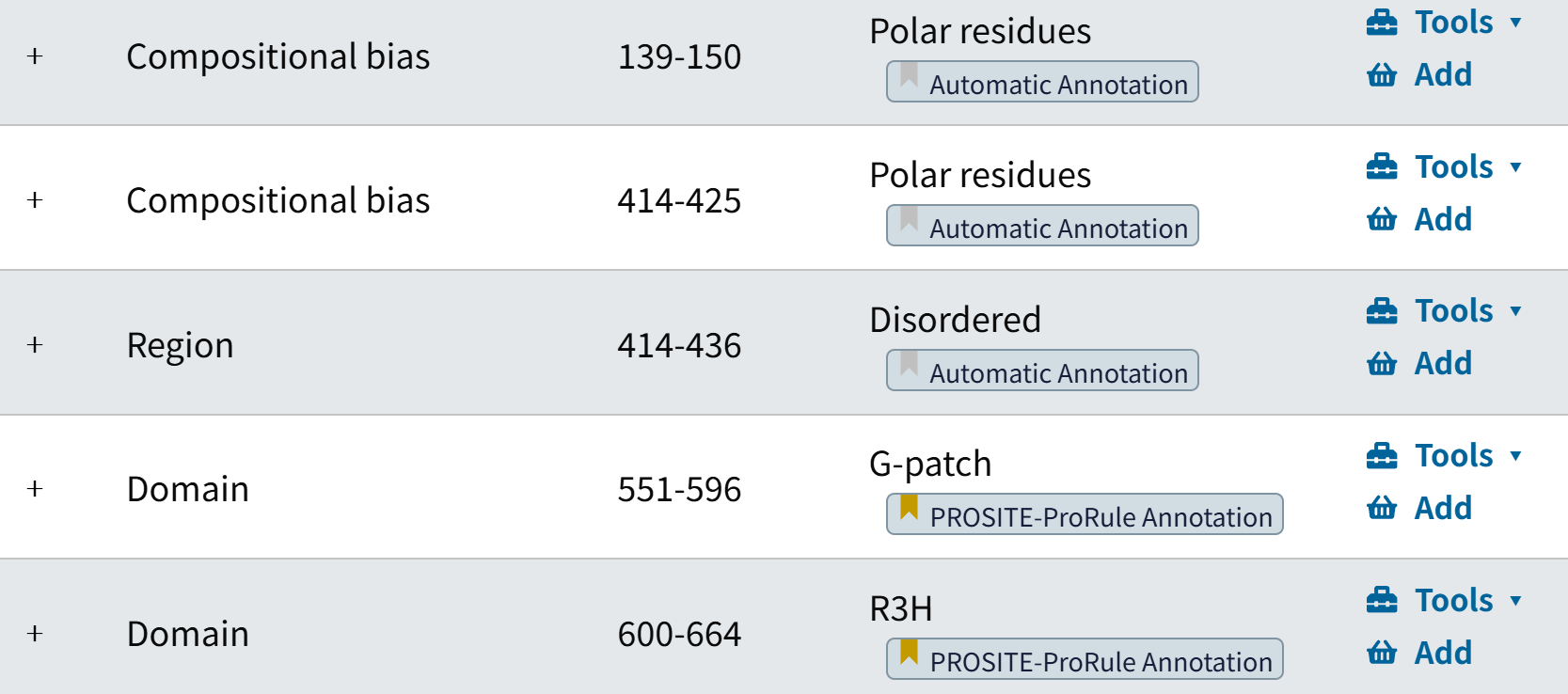
https://www.uniprot.org/uniprotkb/Q14978/entry#family_and_domains
结构域注释不用看,region也不用说,其实主要是看注释
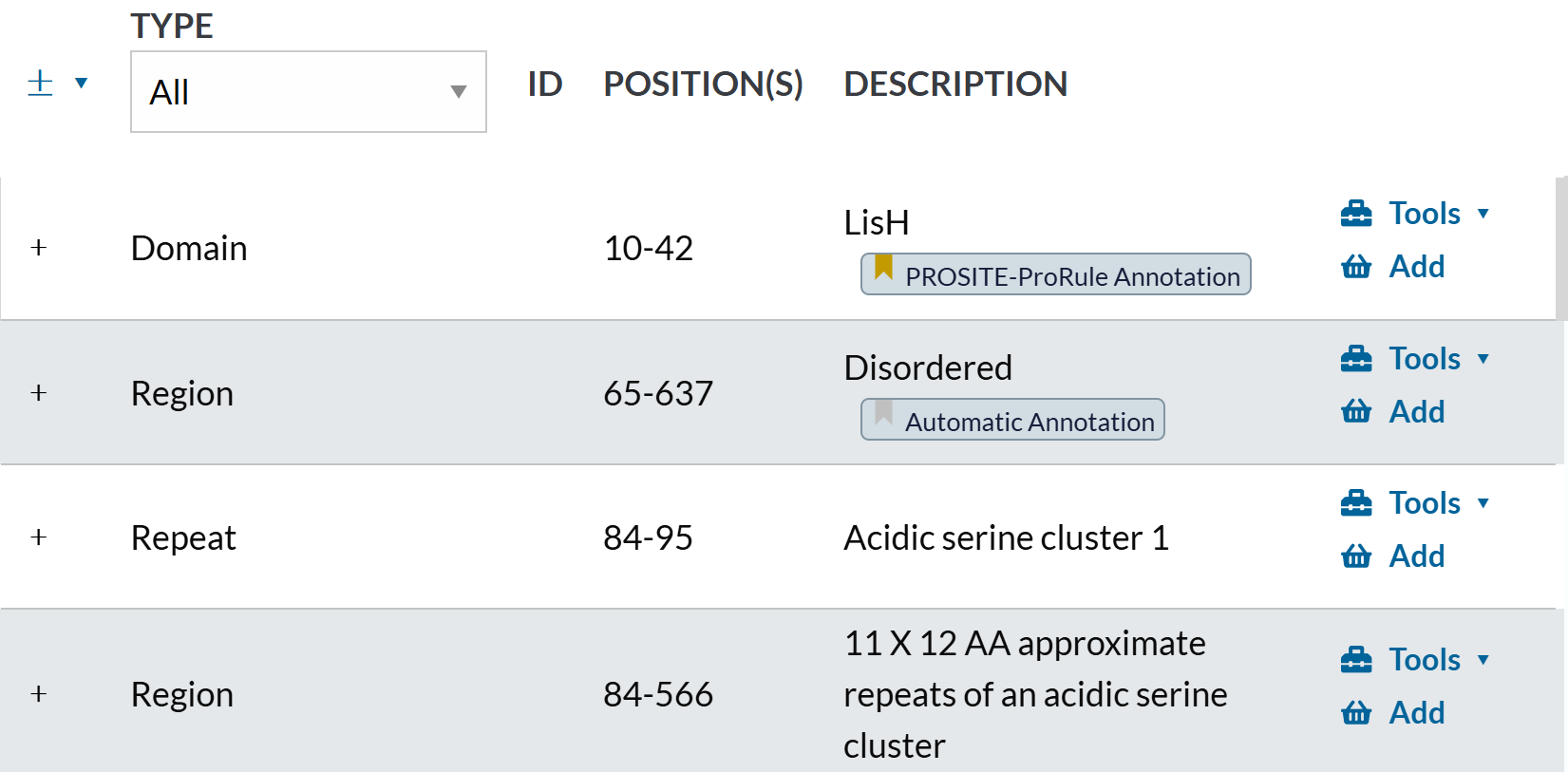
其实类型就那么几个,主要是看其如何描述,
比如说组成偏差上,这里还可以分出酸性残疾富集偏差还是低复杂区域,其实前面说了,这些概念都可以和IDR区域归并在一起;

另外就是Repeat区域,不过这里的数据也同样没有注释,
应该也是单纯序列分析或者是文献中发现注释的。
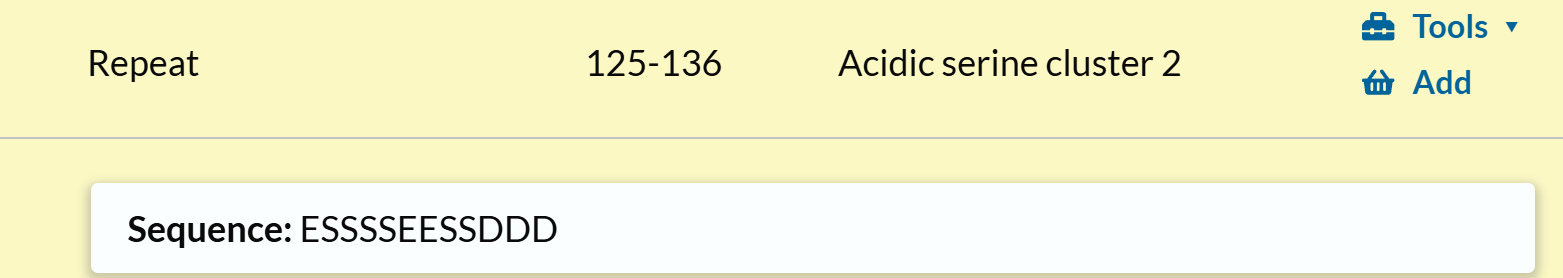
剩下的几个示例条目其实都差不多:
https://www.uniprot.org/uniprotkb/O08919/entry#family_and_domains
https://www.uniprot.org/uniprotkb/Q96L73/entry#family_and_domains
对于结构域的自动注释部分:
在描述中会表明是来自于哪一个InterPro数据库的注释

可以看得出来,单纯从我们的数据来说,我们需要的结构域数据其实混杂在一起:
不仅仅是domain,其实region以及motif类型中都有一些是比较划分混乱的概念,比如说某一个蛋白质互作motif,或者是核定位信号,其实都可以理解为我们在功能分析中所需要的结构域domain数据。
总体类型上大概就下面那么几类:

其中domain的链接参考:https://www.uniprot.org/help/domain,基本上就是上面的这些内容。
然后是motif的参考链接:

这里还是给出两个示例:
https://www.uniprot.org/uniprotkb/Q9FFX4/entry#family_and_domains
一个转录抑制的motif,但还是没有具体的注释来源
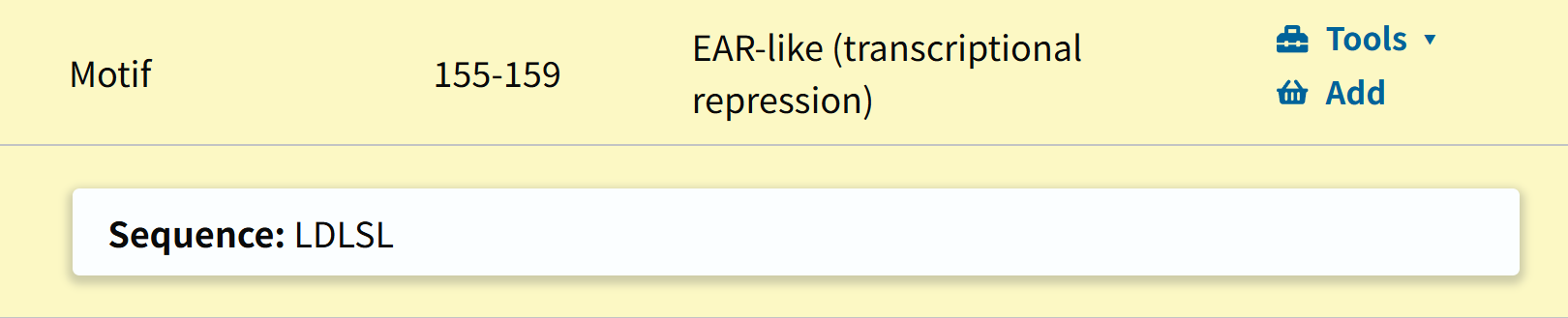
https://www.uniprot.org/uniprotkb/Q8K3W3/entry#family_and_domains
核定位信号:注释来源是序列分析
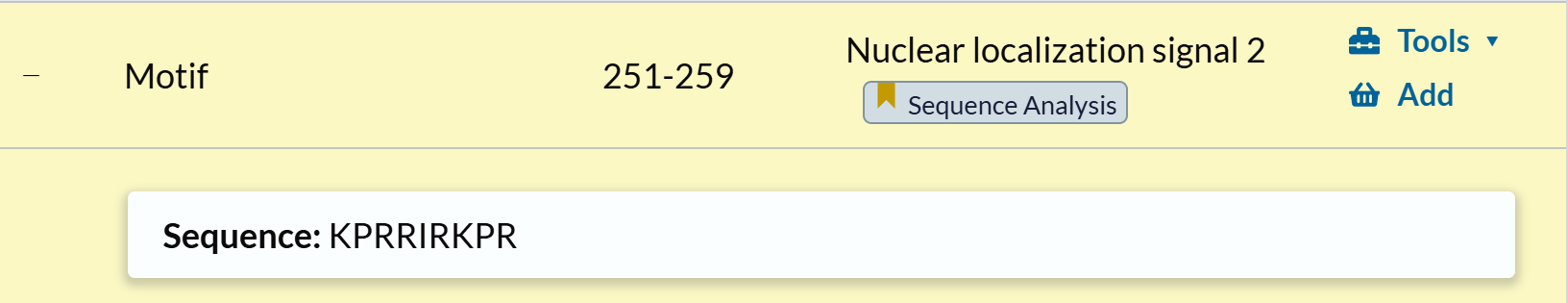
而且是依据序列分析自动断言



Region的参考链接:https://www.uniprot.org/help/region

我们同样来看一下这些示例:
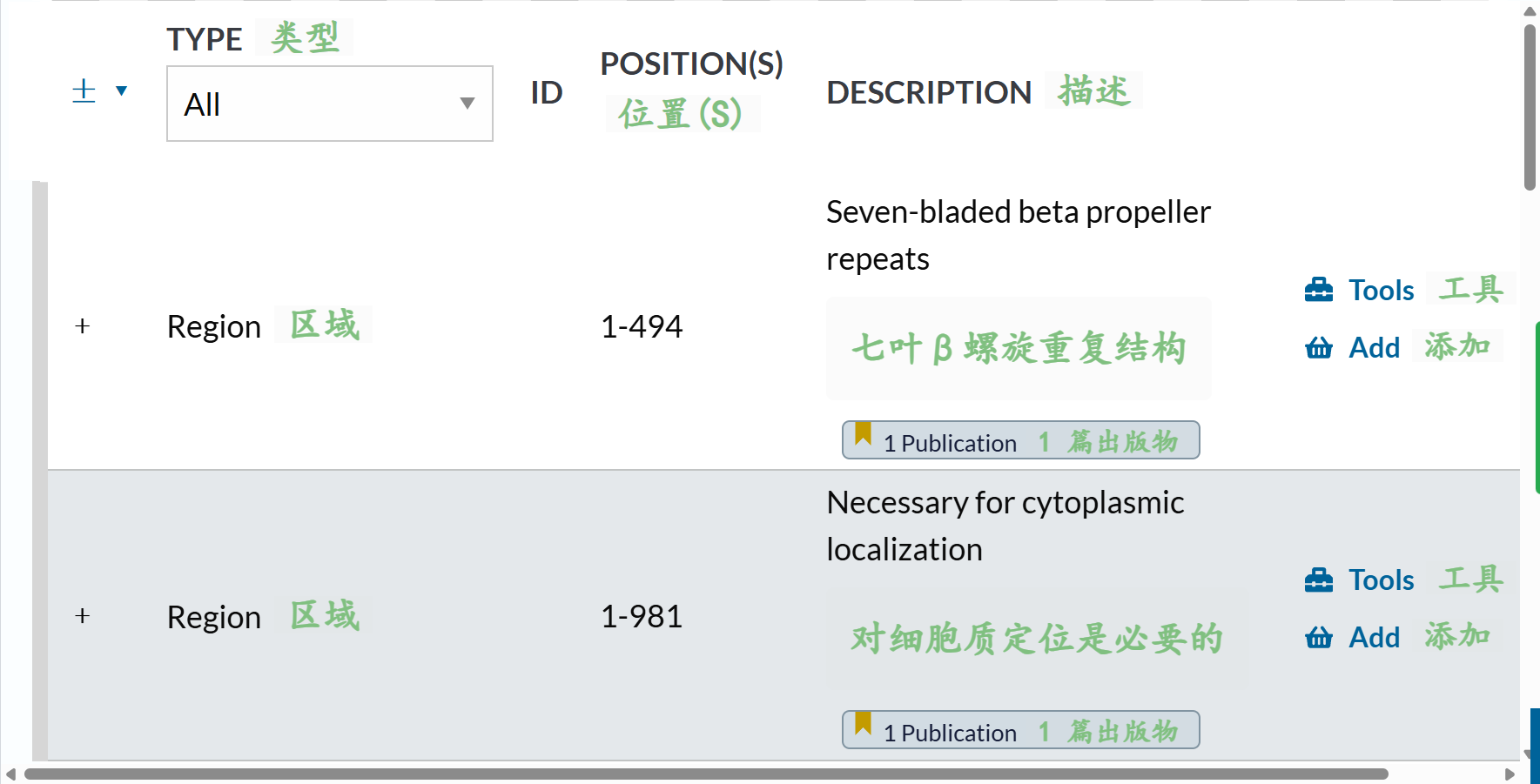
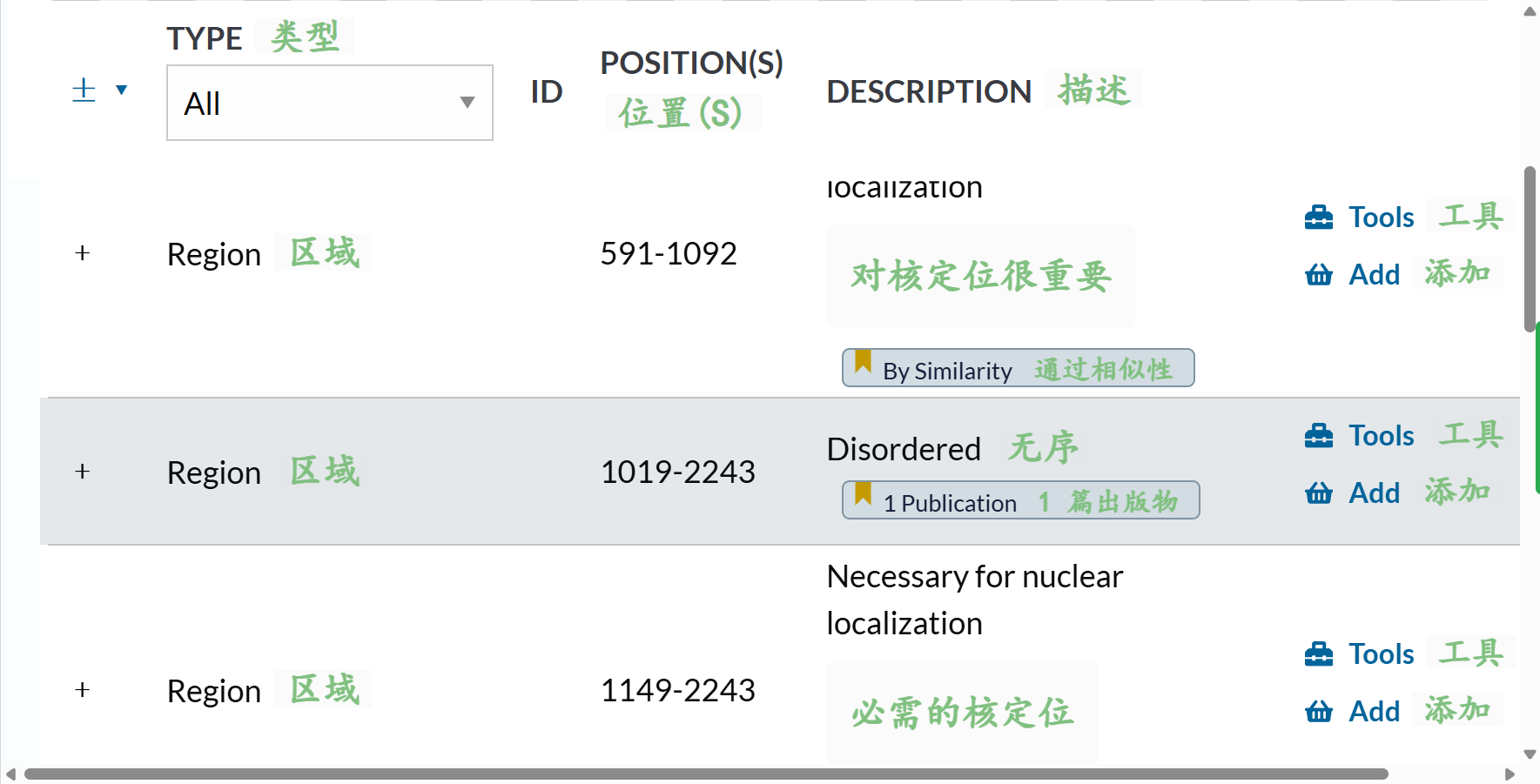

可以发现都是一些很GO式的表述,也就是很文献式的表述,只能说这几个feature之间的表述,确实分类比较混乱;
另外我们也看到了有些disorder的注释,其实并不是数据库注释,比如说uniprot习惯于使用那个我觉得不好的MobiDB-lite,但是我们也可以发现有些disorder是使用文献引用注释的:

其他的就不看了,其实我们可以发现:
region就是那些,在排除了结构域之后,那些在文献中、或者其他注释表明能够介导生物学功能(也就是GO含义)的、但是又不是结构域那样固定化定义的区域序列。
总体而言,我个人认为,region大多是指那些能够类似domain一样发挥GO生物学功能,但是又没有domain那么严谨定义的区域。
Repeat的参考链接:https://www.uniprot.org/help/repeat

我们可以发现,这个定义就是和前面的几个概念胡在一起的。


https://www.uniprot.org/uniprotkb/P15305/entry#family_and_domains
下面这个才是我们正常理解中的repeat:
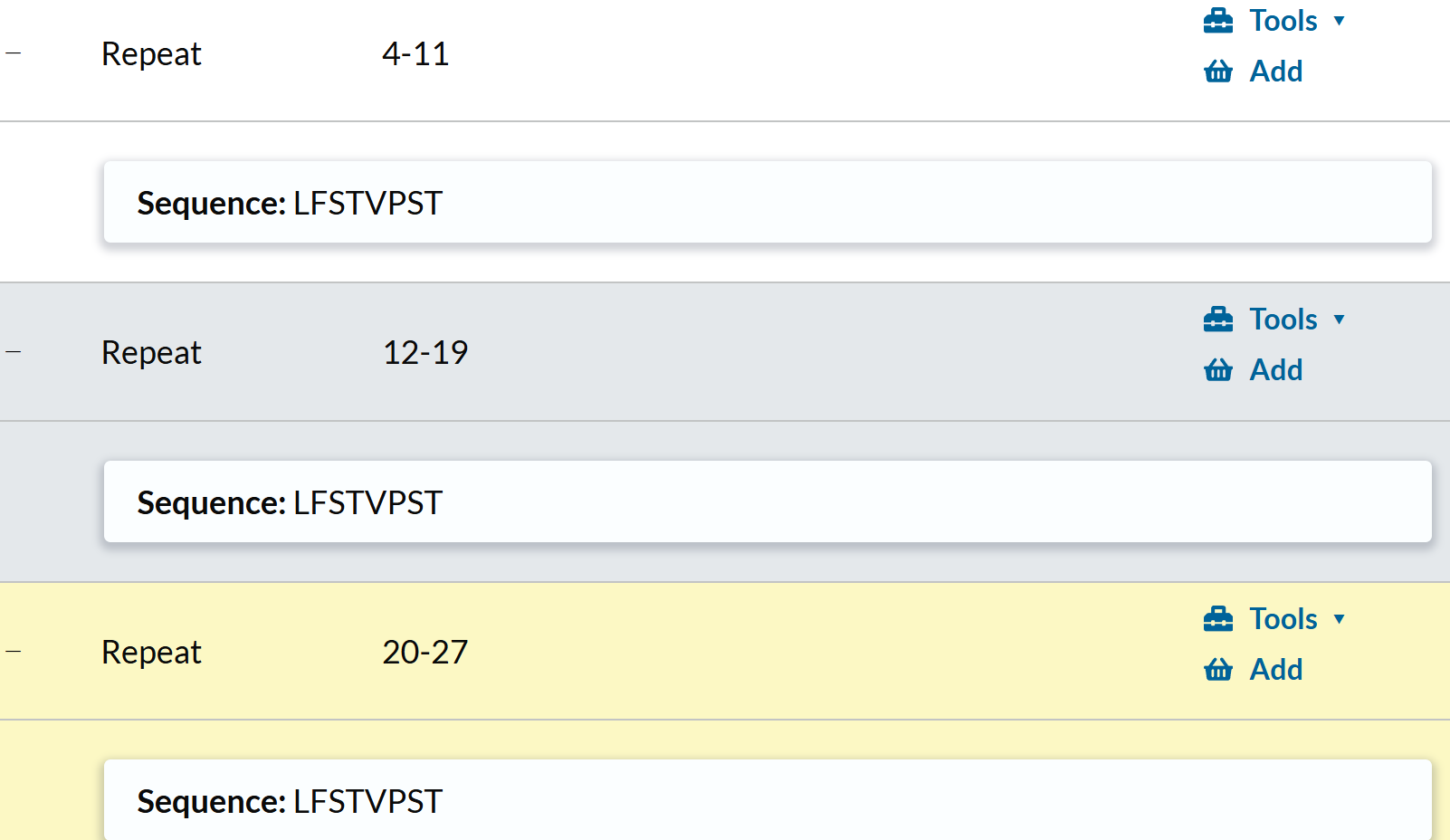
https://www.uniprot.org/uniprotkb/P38479/entry#family_and_domains
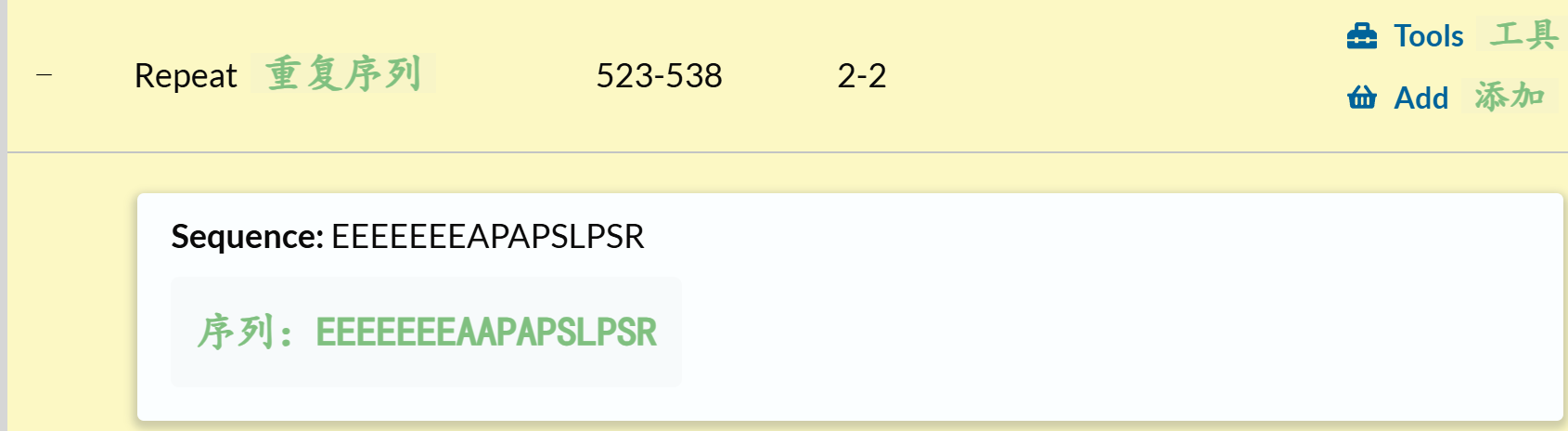

其余的注释条目:
https://www.uniprot.org/help/act_site
与酶相关,而且是残基,per residue,不是region



https://www.uniprot.org/help/var_seq
这个其实就是isoform,打开几个示例链接都是这样,基本上不是在结构域注释数据中,可以pass;


https://www.uniprot.org/help/binding
结合其实是一个生物学事件,算是一个GO中的BP,所以其实这个算是BP类型的数据




binding一般不在界面中的结构域注释部分,所以我们不一定要保留;
https://www.uniprot.org/help/chain
可以发现是成熟蛋白质形成的范围,不是结构域相关信息

打开示例链接,其实这个chain是在PTM section中的:
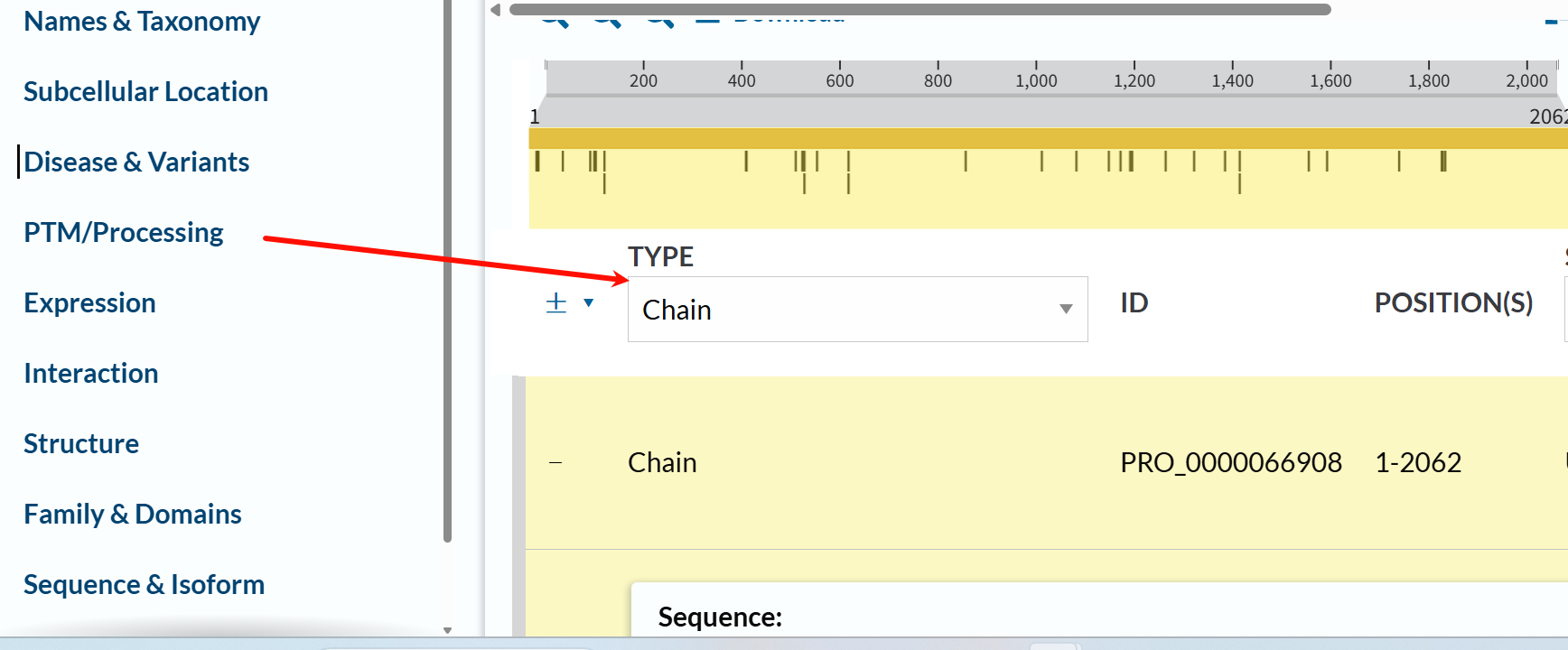

https://www.uniprot.org/help/coiled



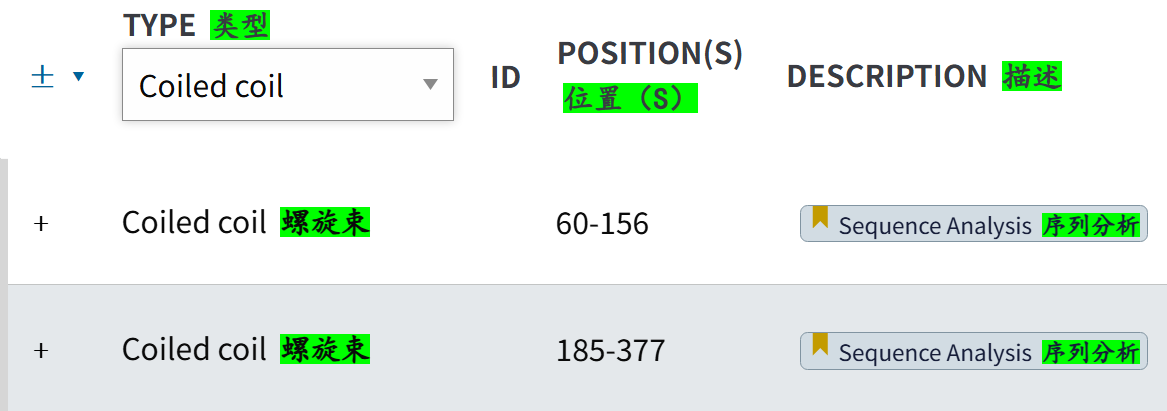
随便看一个示例,这个确实是在domain section中的,
而且其实看概念,螺旋束应该是二级结构α-螺旋的一个集合,也就是超二级结构之类,所以很难说这是一个domain概念,只能说是一个结构概念,但是uniprot数据库中确实又是将这个注释类型放在domain中的,所以我们也可以放进我们的domain研究目标中。
http://uniprot.org/help/compbias
这个也是在domain section中的(也就是family&domain),而且注释使用的是MobiDB-lite,但是我们知道这个数据库算法其实是用来注释IDR的,
所以可想而知,如果有IDR 注释,基本上大多数会和这个bias重叠;
而且MobiDB-lite的注释算法其实我个人觉得很落伍了,至少在CAID比赛上,benchmark上基本上没怎么看到这个工具,大概表现不尽人意,被其他深度学习+生物物理建模的工具压下去了。


随便打开一个示例:
有LCR重叠的

也有这种示意的:

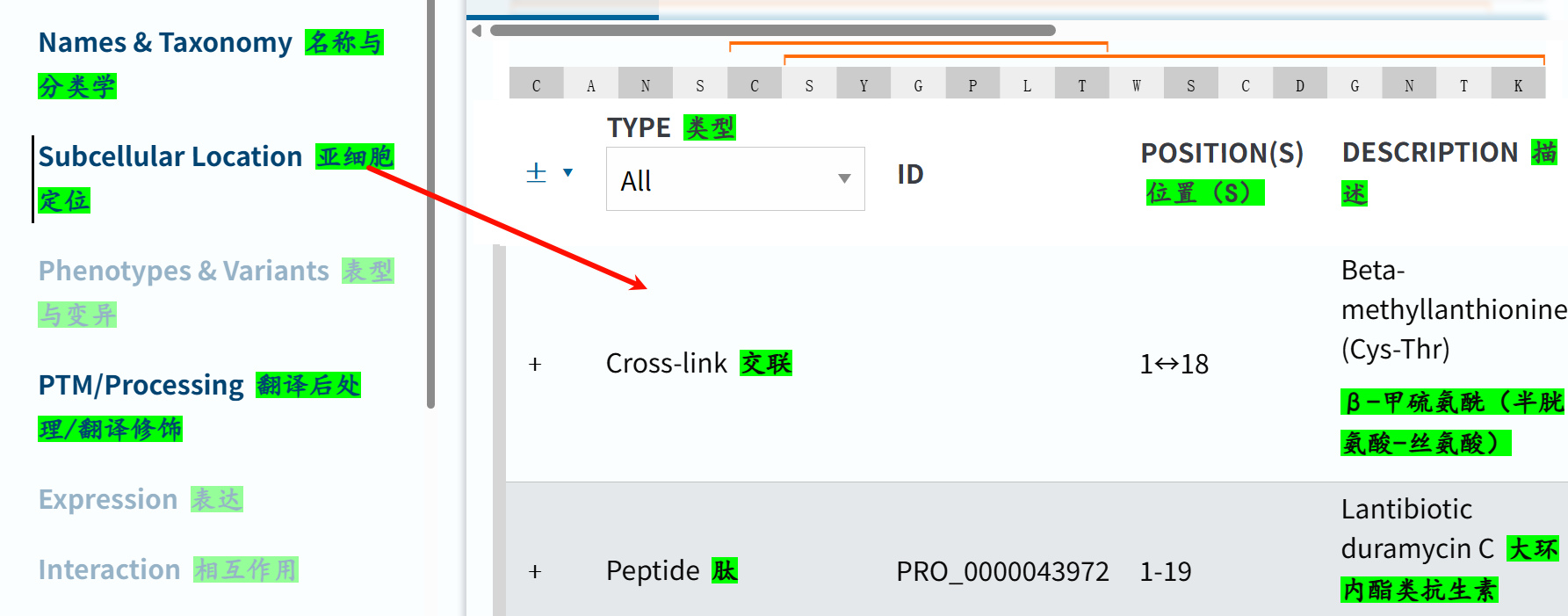
https://www.uniprot.org/help/crosslnk
广义上来讲就是翻译后修饰,也就是蛋白质成熟过程中的处理



这个其实一看就是在PTM模块中的,所以也不算是processing












还有很多文档我就不一一展开描述了
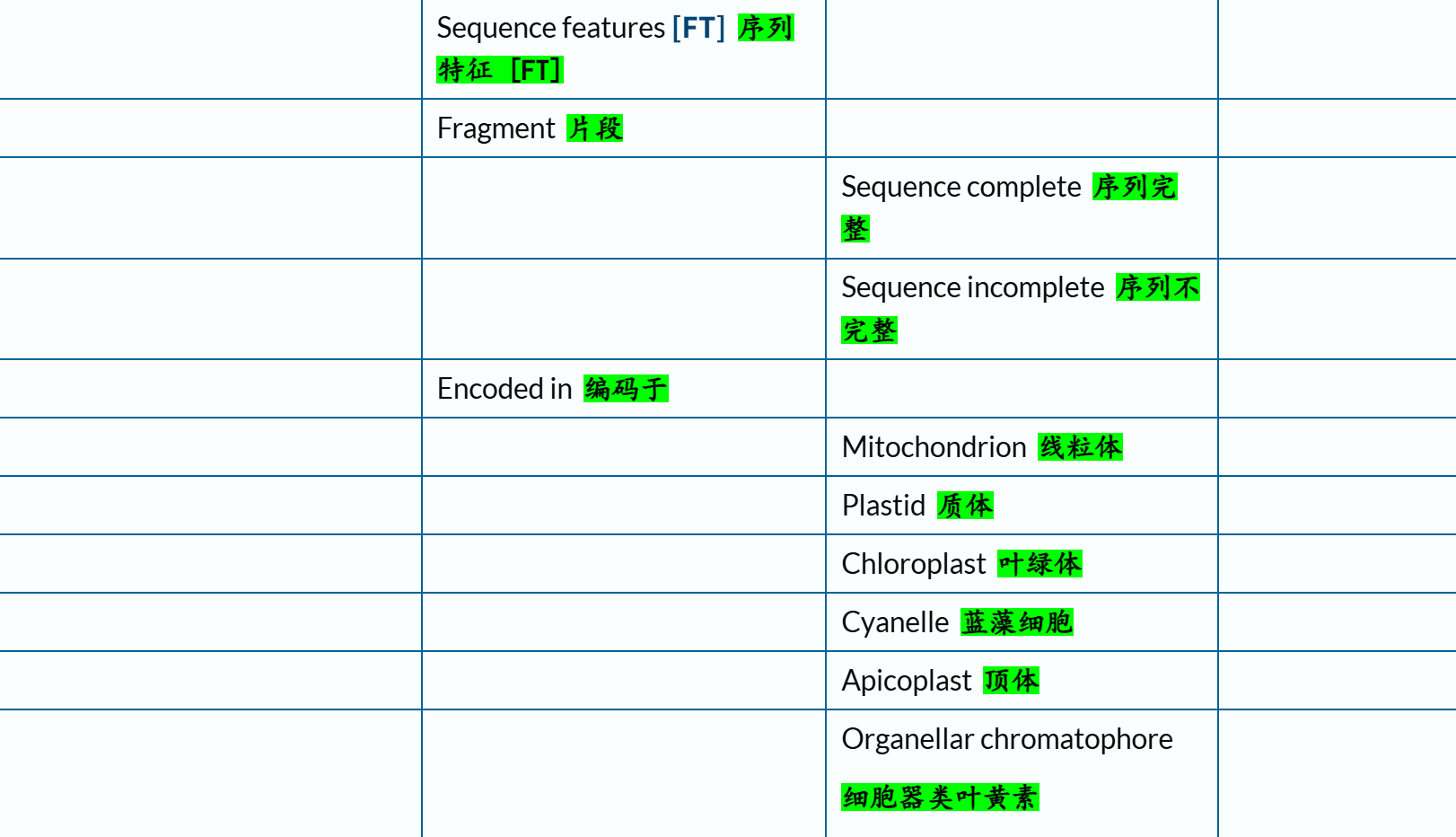
总之,参考了上述官方文档之后,我个人是倾向于保留下面这几个条目,用绿色✅标注(标注👍的则是下文再用代码示例验证之后):
# Family and domains 偏向于结构域
'Coiled coil' # 多条α-螺旋形成的超螺旋, 结构概念 ✅👍
'Compositional bias' # 组成偏倚或过表达aa bias区域, 会和IDR概念重合 ✅👍
'Domain' # 二级结构组合, 具三级结构, 普遍意义 ✅👍
'Motif' # 保守序列, 类似于核定位信号之类 ✅👍
'Region' # 实验上定义的区域特征, 感兴趣的区域 ✅👍(但需要排除IDR)
'Repeat' # 蛋白质内部重复序列基序或重复结构域的位置和类型 ✅
'Zinc finger' # function section, 但是注释在domains中 ✅👍# function
# 参考:https://www.uniprot.org/help/function_section
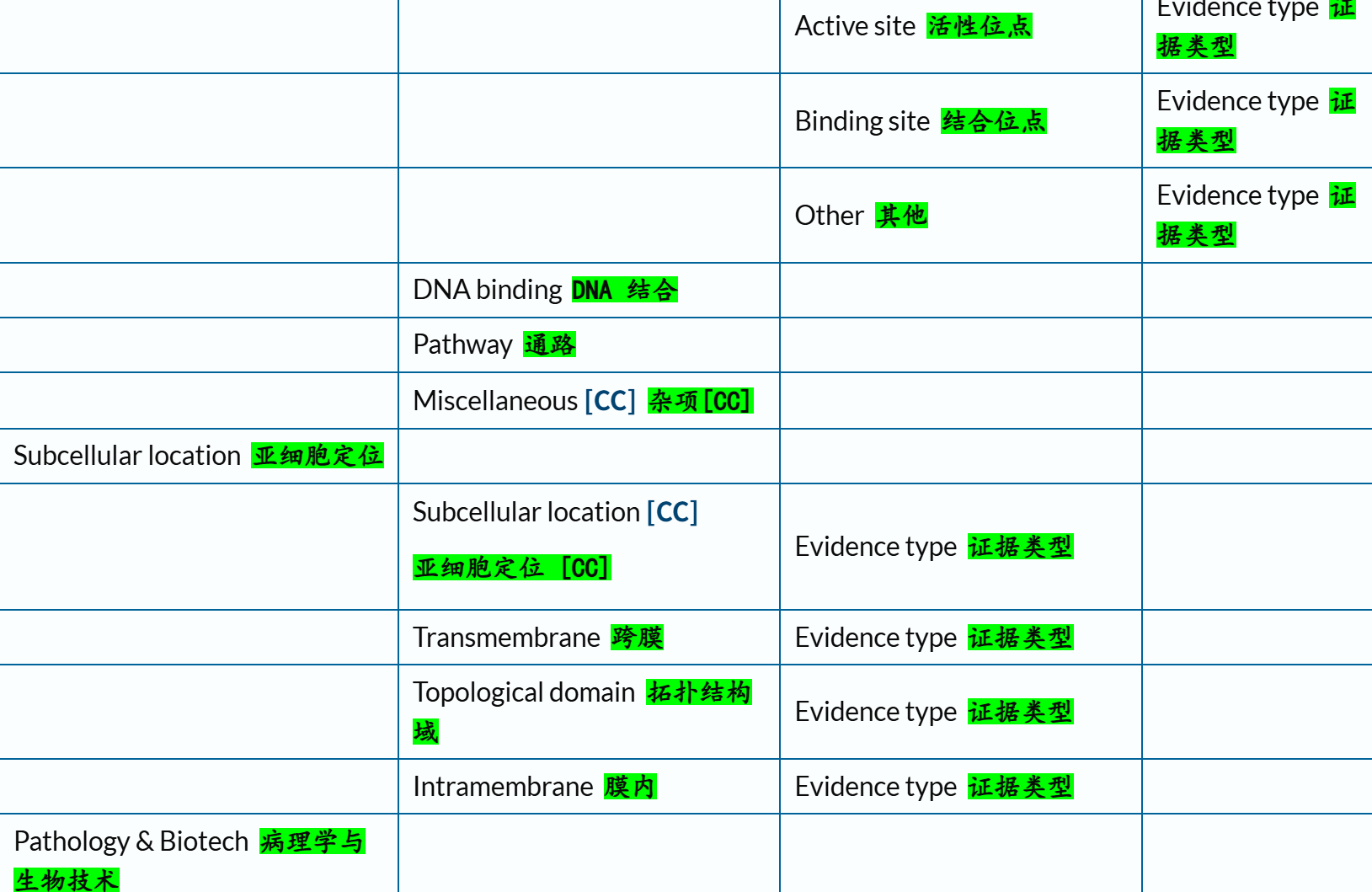
'Active site' # 直接参与酶活性的氨基酸 ✅👍
'Binding site' # 蛋白互作位点 ✅👍
'DNA binding' # DNA结合结构域位点+类型 ✅👍# https://www.uniprot.org/help/site
'Site' # 有趣的单个氨基酸位点 ✅👍# sequence
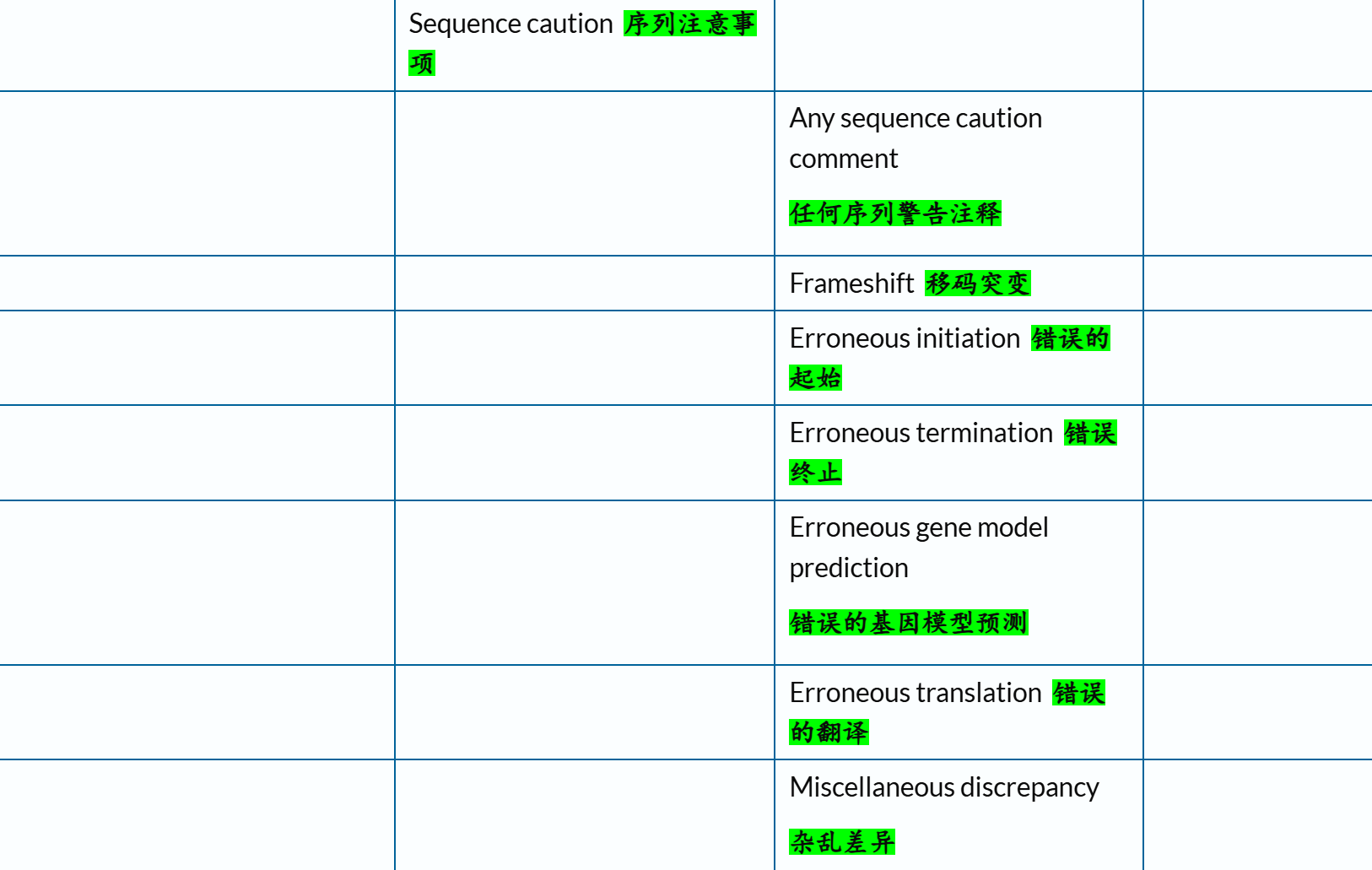
'Alternative sequence' # 异构体isoform
'Sequence conflict' # 提交序列与规范序列不同的位置、由此产生的蛋白质序列变化以及与不同序列报告对应的参考 # structure 结构(低于前面的结构域概念)
'Beta strand'
'Helix'
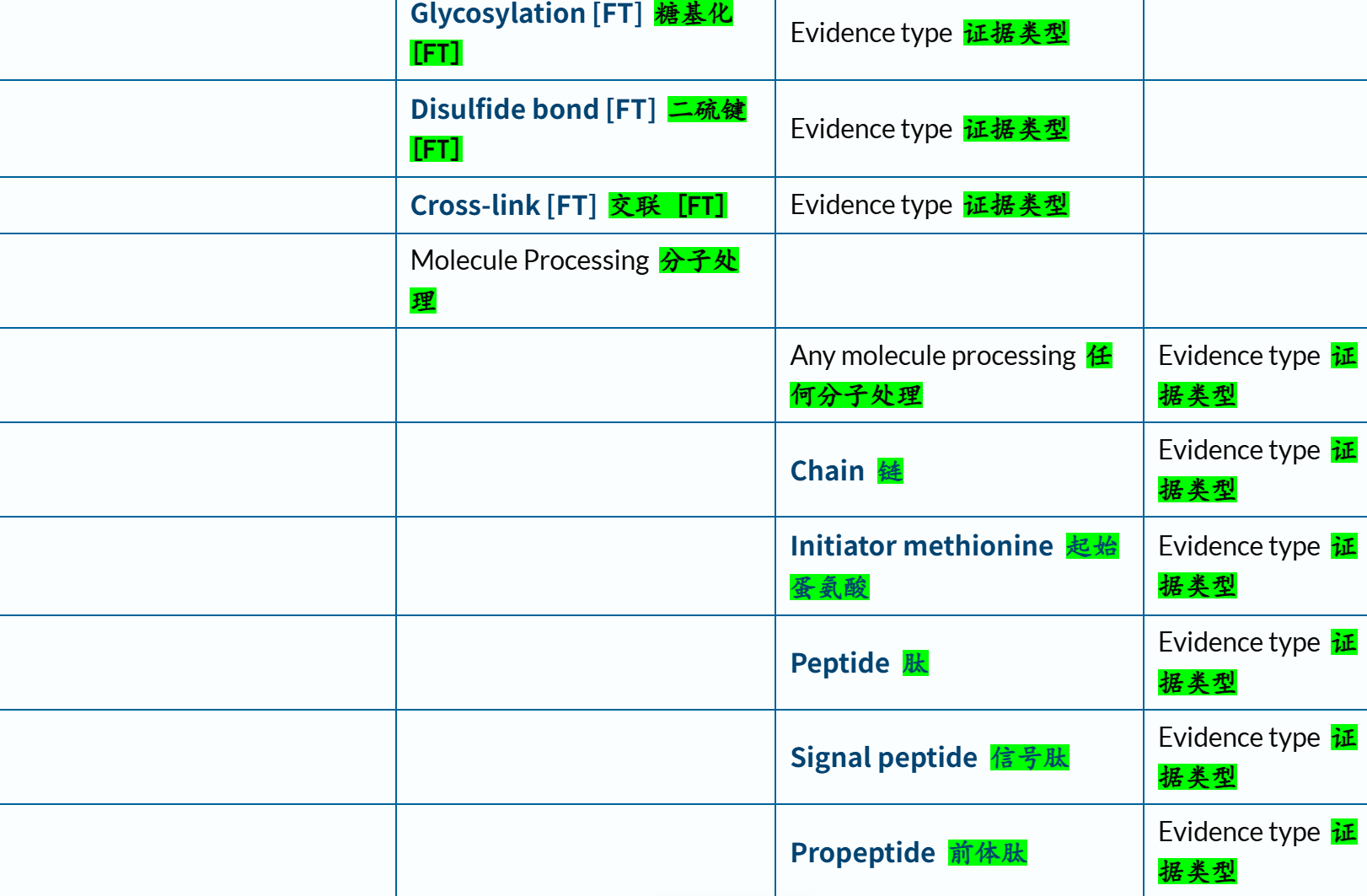
'Turn'# PTM / Processing 成熟蛋白质形成
'Chain' # 多肽链
'Cross-link' # 蛋白质形成过程中的共价连接
'Disulfide bond' # 二硫键, 和上面这个合并在一起
'Glycosylation' # 糖基化, PTM的一种
'Initiator methionine' # 起始蛋氨酸从成熟蛋白质中被切割
'Modified residue' # 除脂质、聚糖和蛋白质交联外,每种修饰残基的位置和类型 ✅👍
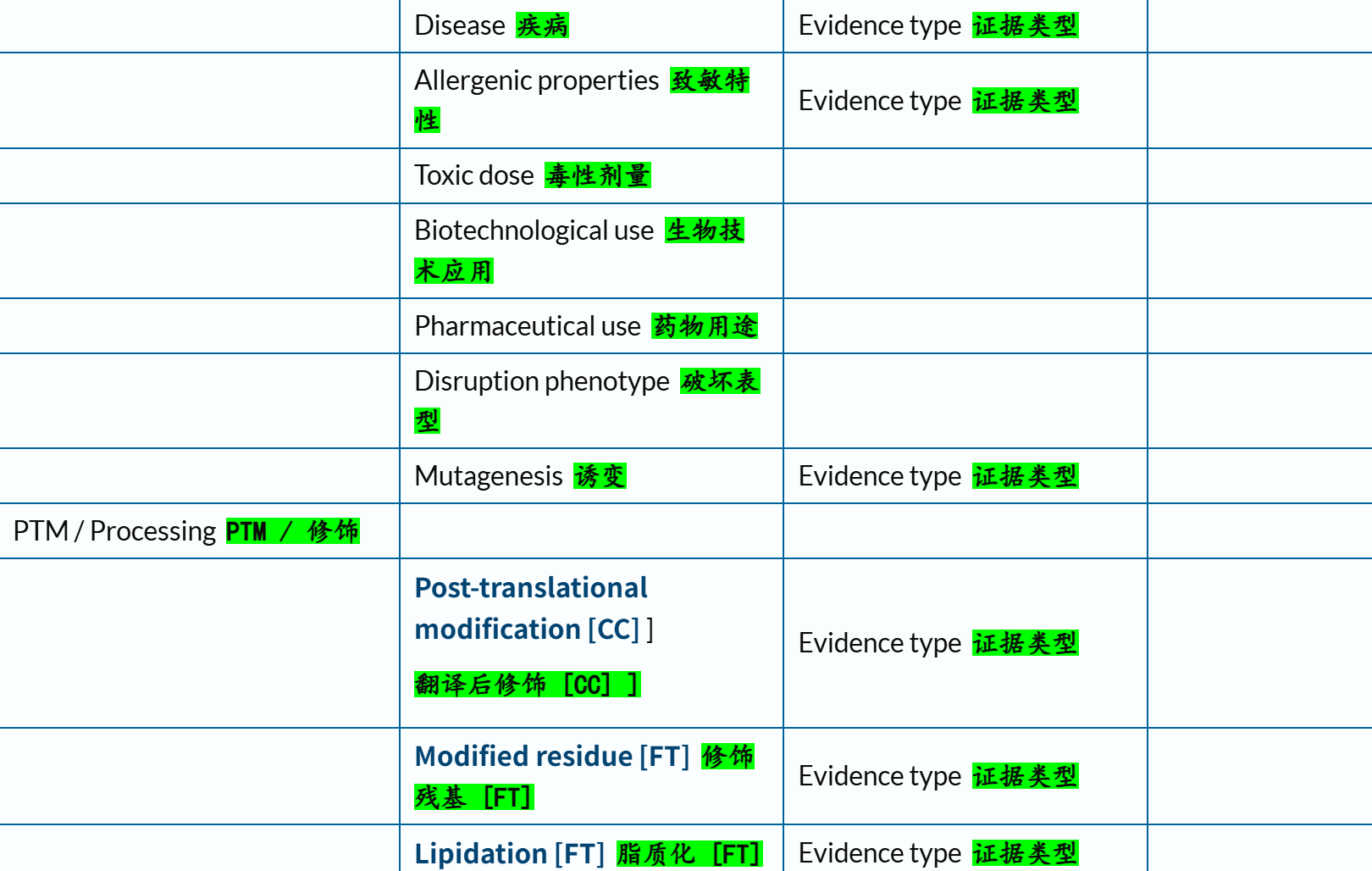
'Propeptide' # 前体肽段, 蛋白质在成熟或激活过程中被切割的一部分。一旦被切割,前体肽通常没有独立的生物学功能# Disease/Phenotypes and variants 疾病与表型变异部分
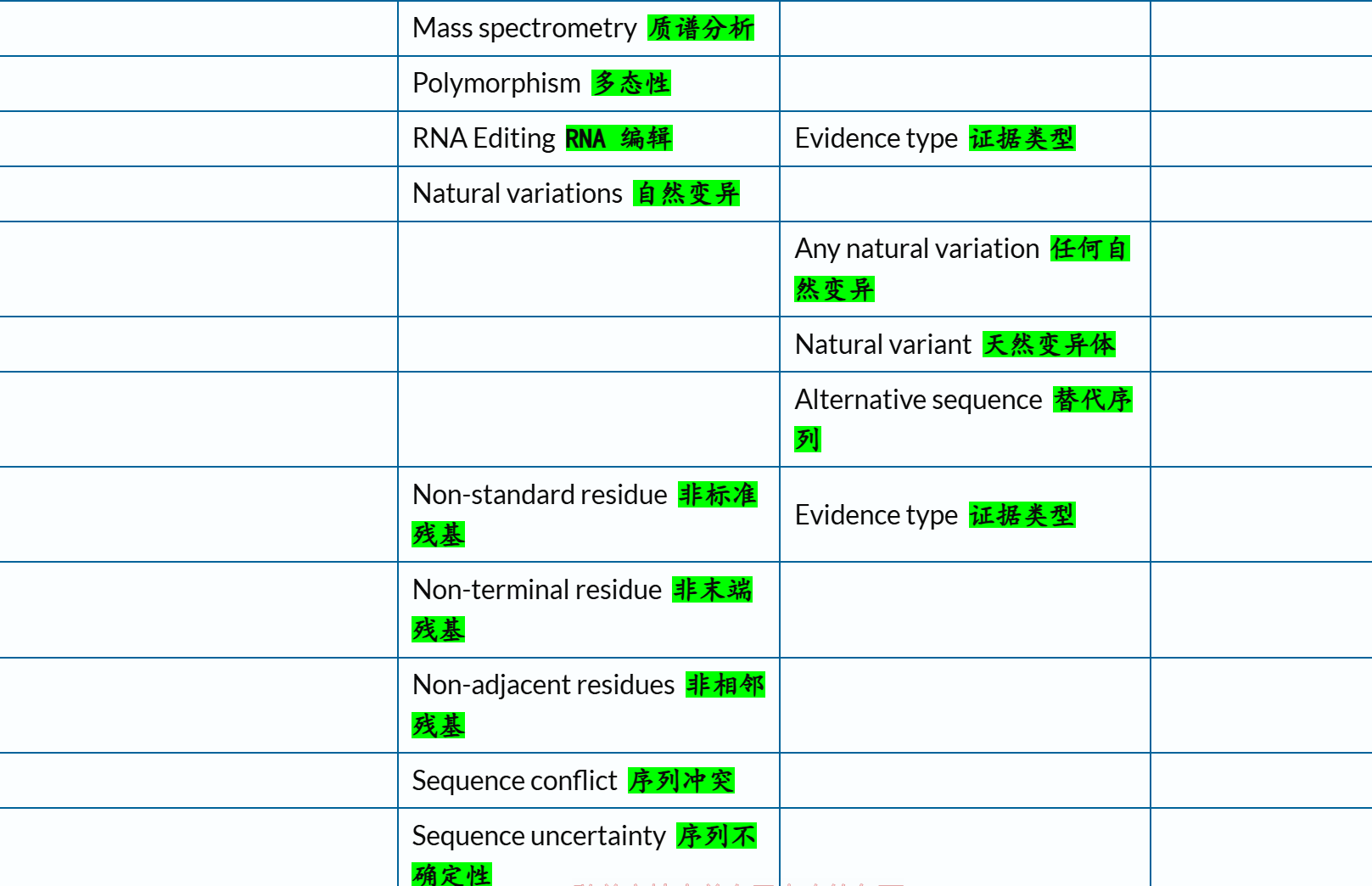
# https://www.uniprot.org/help/disease_phenotypes_variants_section
'Mutagenesis' # 位点突变数据 ✅👍
'Natural variant' # 蛋白质序列的自然变异
看起来还有十几个,可能数据还是很多,我们要结构域没必要要这么多,我们可以再次使用代码来处理一下
import requests,time
# 使用stream接口获取数据
url = "https://rest.uniprot.org/uniprotkb/stream"query = "(organism_id:9606) AND (reviewed:true) AND ((xref:prosite-PS00028) OR (xref:prosite-PS50157))"headers = {"Accept": "application/json"}params = {}
params['query'] = queryresponse = requests.get(url, headers=headers, stream=True, params=params)data = response.json() fea_type_list = []
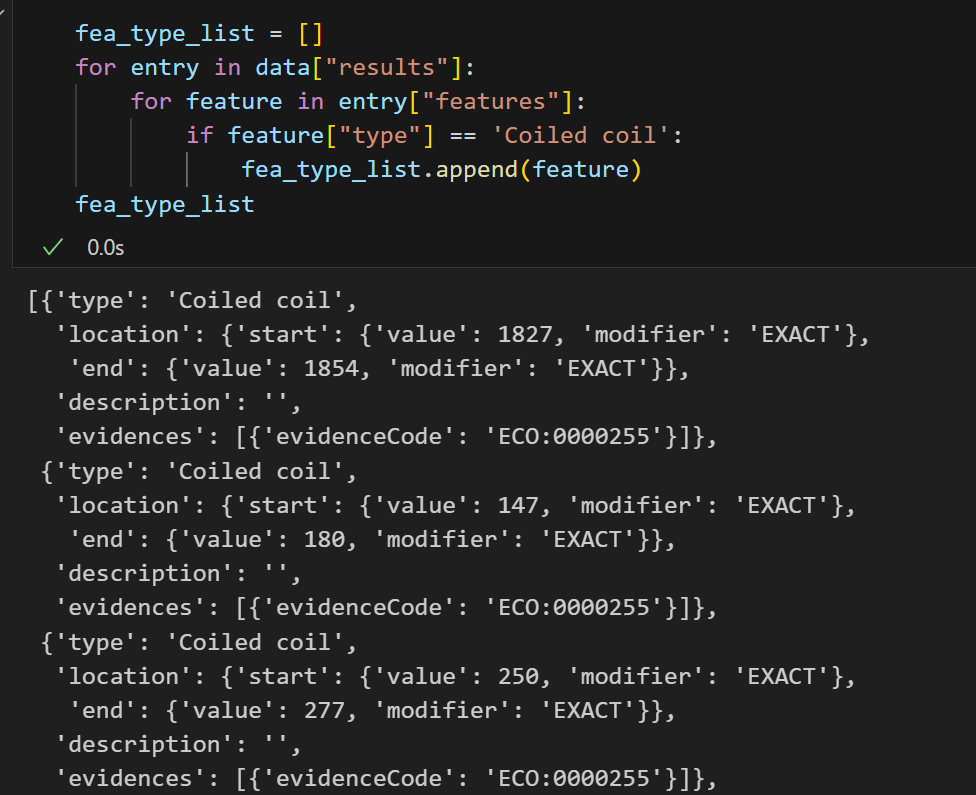
for entry in data["results"]:for feature in entry["features"]:if feature["type"] == 'Coiled coil':fea_type_list.append(feature)
fea_type_list
这个是超螺旋,我看了下,还是觉得挺好,留一个
组成偏倚,看着还可以
domain,不用说,肯定是要的
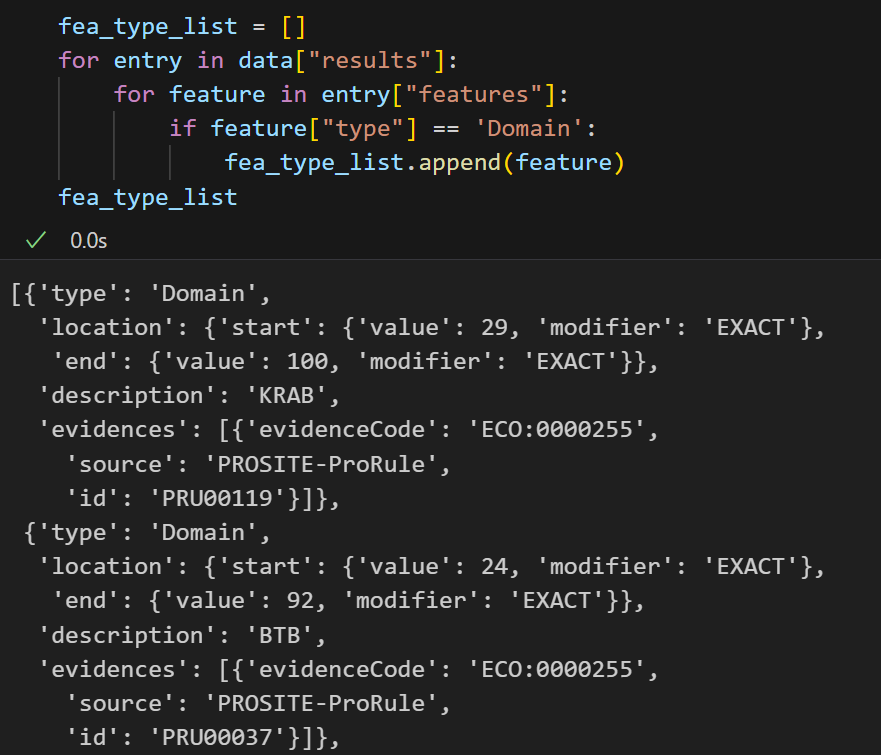
motif也还可以:
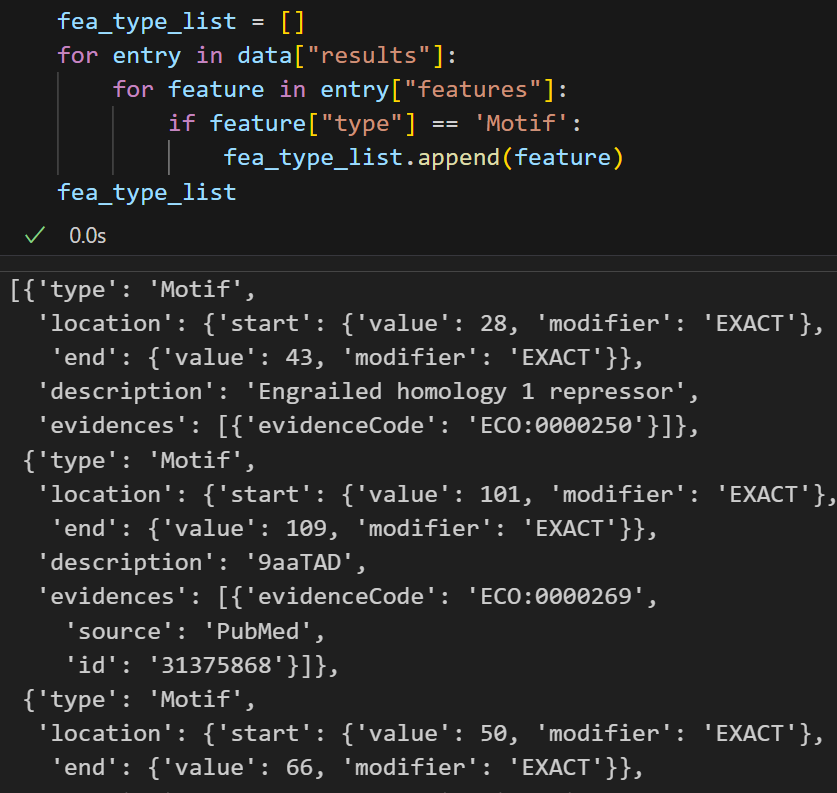
Region还可以,但是我看大多数都是描述disorder区域,但是我IDR区域完全可以用更好的工具来注释,所以这里Region可以保留,但是要排除掉’description’: 'Disordered’的项目
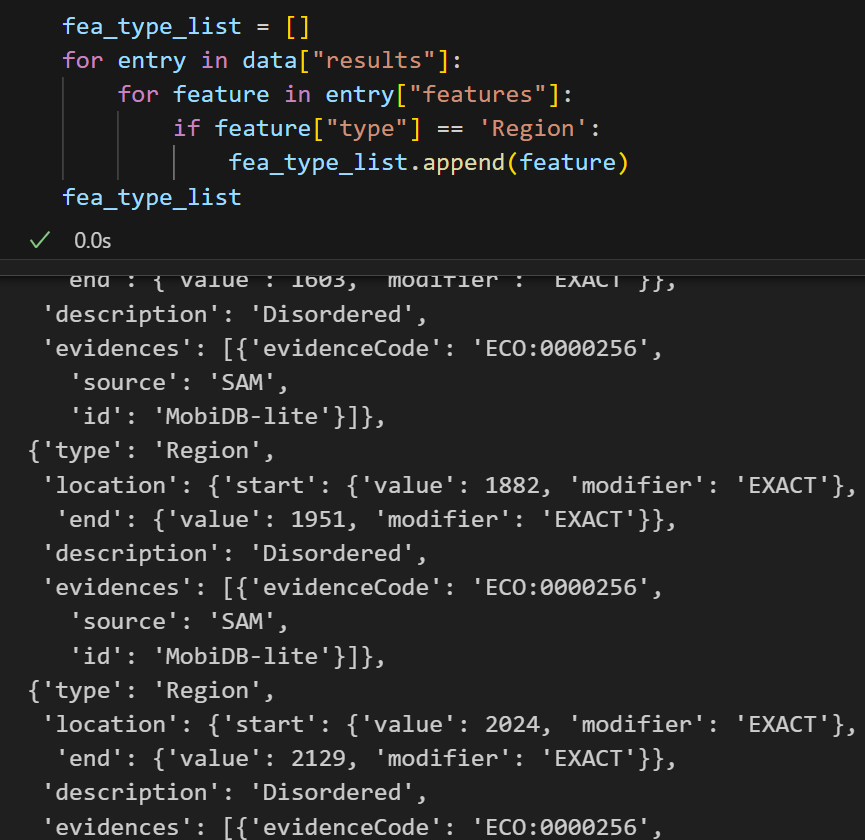
Repeat看着就比较拉了,都是一些无聊的数据
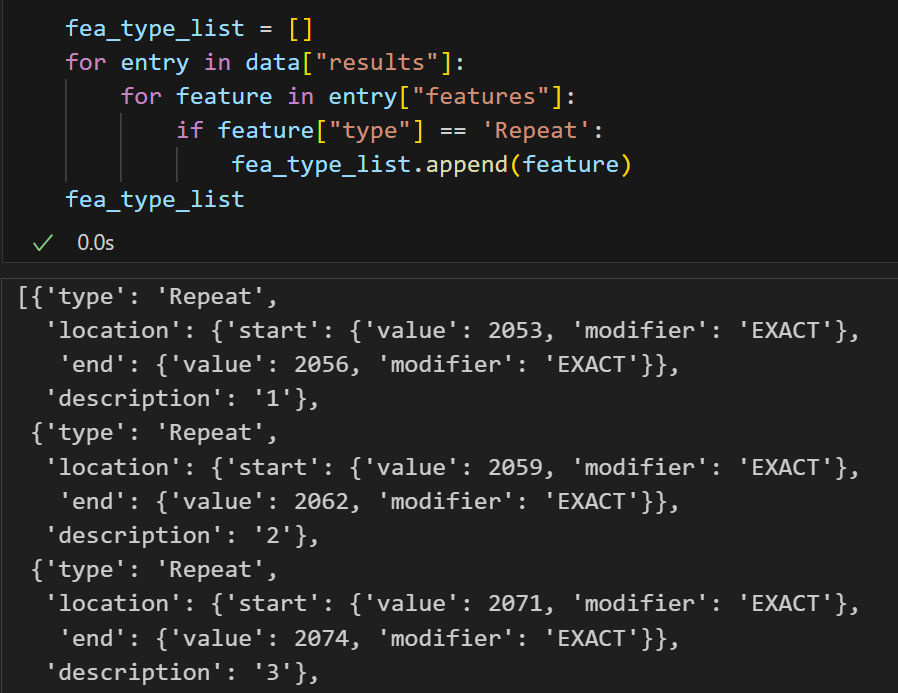
锌指也没得说:
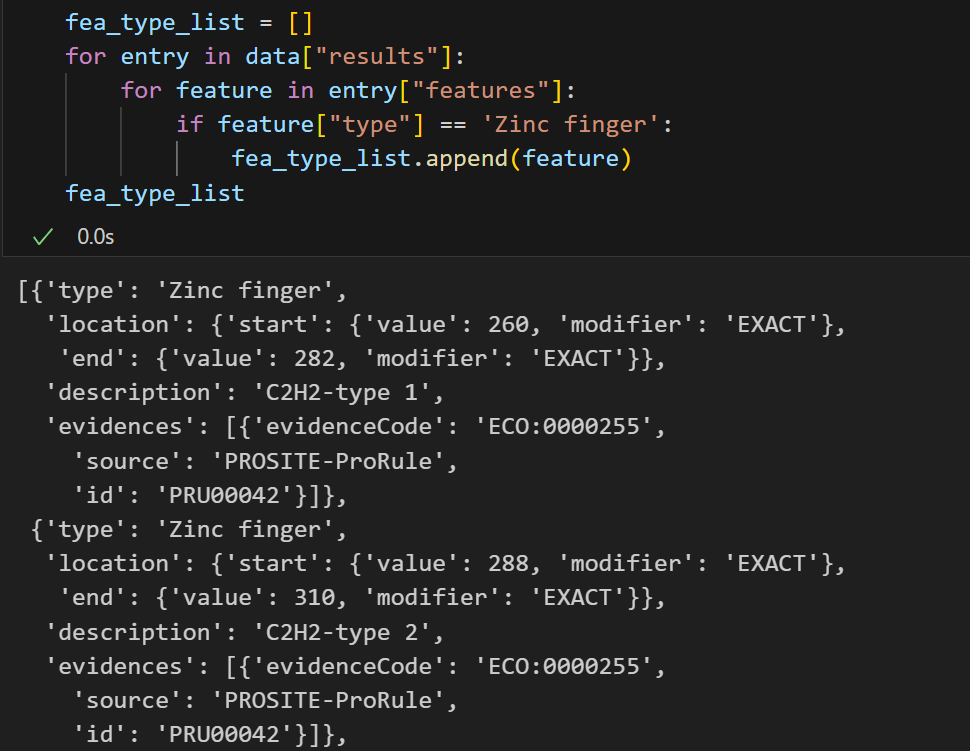
酶活性位点,非常少,感觉没意思,不保留这个数据
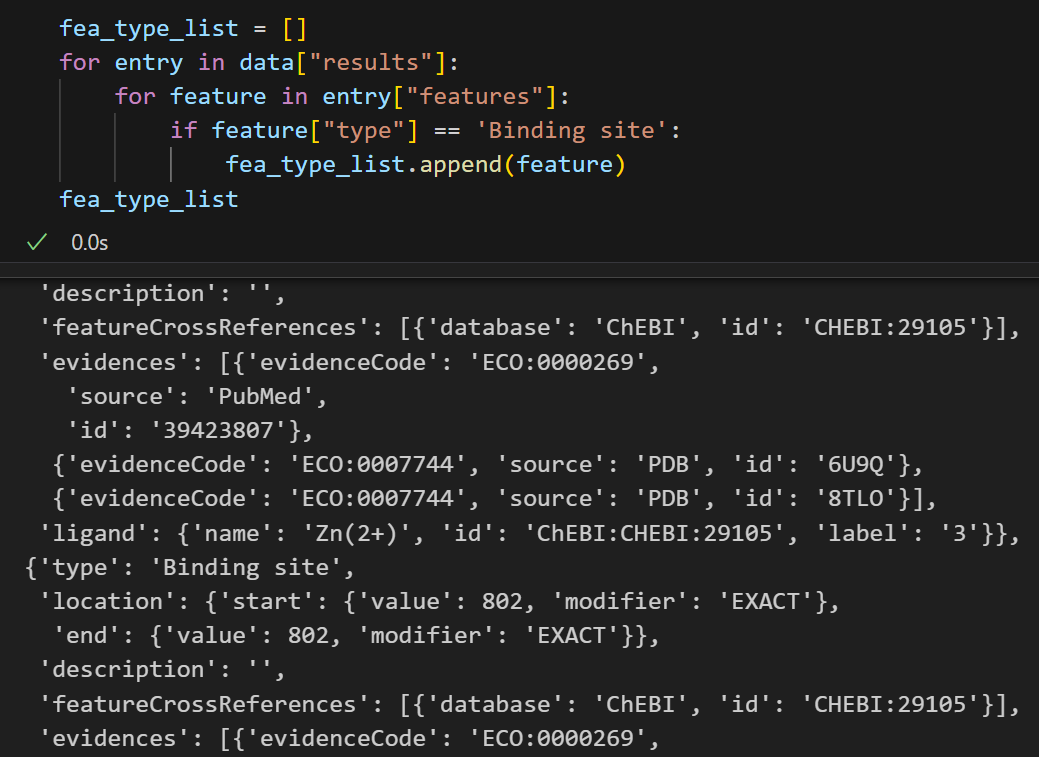
binding site这个数据有点尴尬了,有结果也不知道怎么解读提取
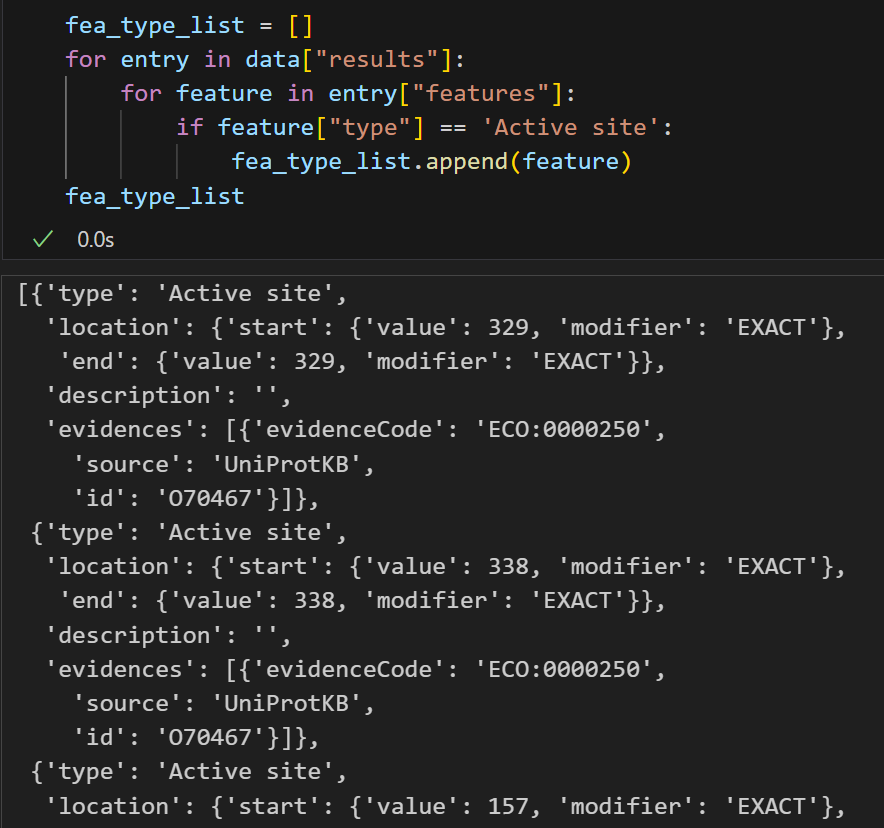
这个数据也挺尴尬的,很少,大多数甚至都没有DNA binding,简直无法想象,还是保留
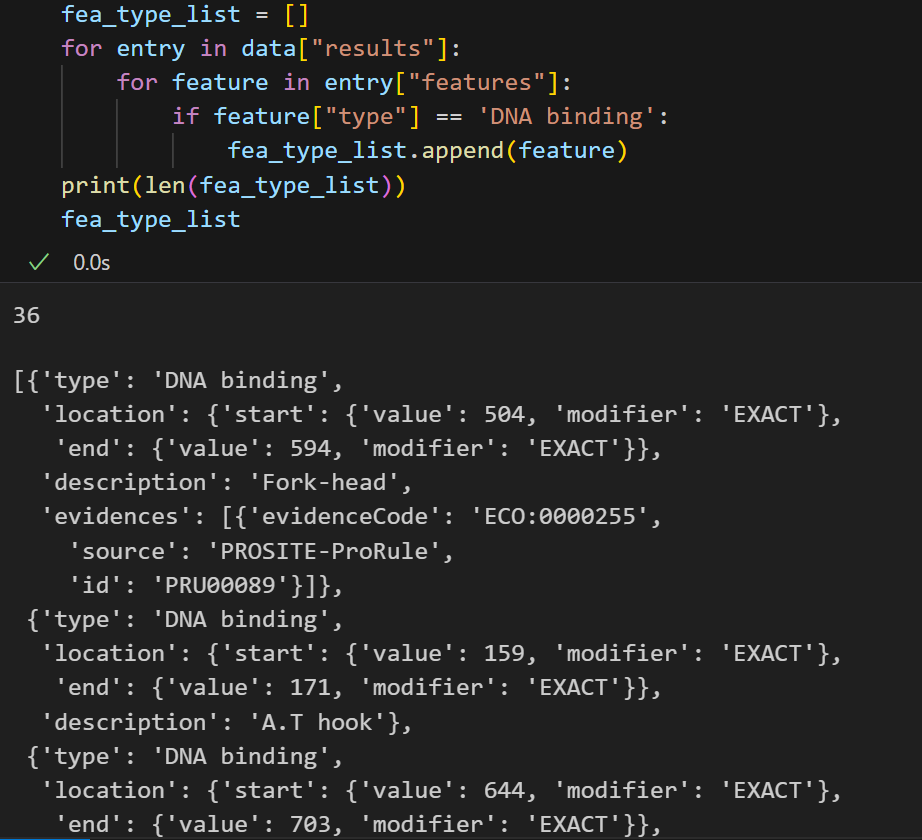
翻译后修饰,可以保留
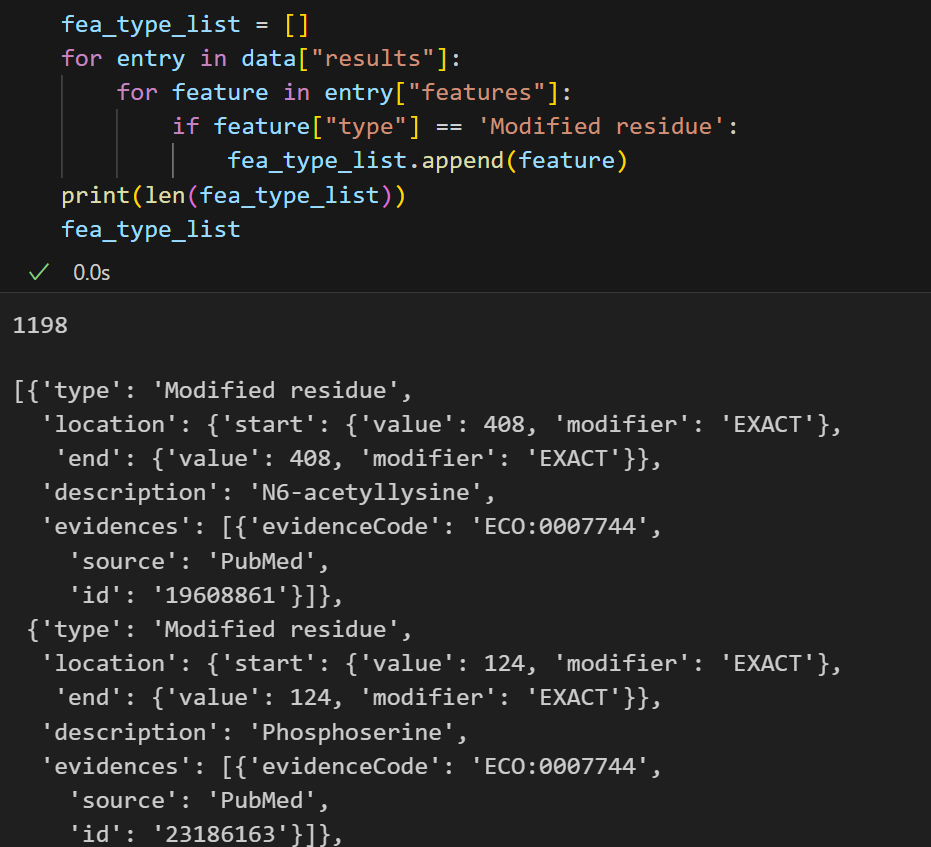
突变位点数据,可以保留
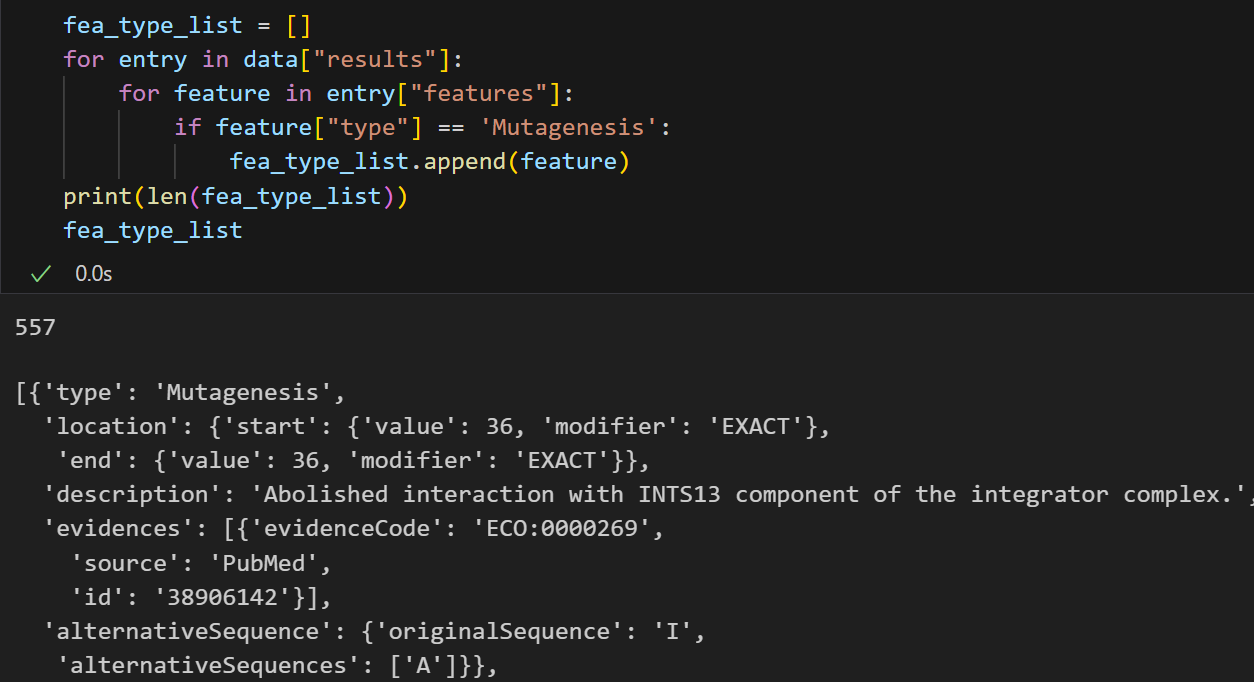
位点,还行
其实分析到这里,我的选择困难症又犯了,所以一个比较折中的方法就是:
保留所有的features类型数据,增加一个key,也就是保存为dict类型数据,每一个key是描述或类型,然后value可以使用dataclass保存为一些复合型的数据,
对于dataclass,可以参考之前OOP部分的博客:https://blog.csdn.net/weixin_62528784/article/details/152951342?spm=1001.2014.3001.5501
现在我们开始来写一个获取所有feature类型的脚本,所有feature都能获取了,其实结构域数据就是只抓取其中某些feature类型,逻辑其实是一样的。
假设我们已经获取了某个蛋白质id,还是以CTCF为例子:
我首先使用dataclass数据结构来存储我待会分析的输出,之所以我只用type、description、location(start,end)这几个键值对来存储数据,是因为我提前看了一下我query过的数据,大概这些键值对都有,而且我基本上只看feature中的这些部分。
参考:https://github.com/MaybeBio/bioinfor_script_modules/blob/main/41_Get_Protein_Annotation.py
from dataclasses import dataclass
from typing import List, Dict, Optional, Tuple
import requests,time@dataclass(frozen=True)
class UniProt_Feature:"""Description----------UniProt特征类, 存储从UniProt数据库获取的蛋白质特征信息Args----------feature_type (str): 特征类型, 例如'DOMAIN','REGION','MOTIF'等description (str): 特征描述信息start (int): 特征起始位置(1-based)end (int): 特征结束位置(1-based)"""feature_type: strstart: intend: intdescription: Optional[str] = None@dataclass(frozen=True)
class UniProt_Feature_Analysis_Result:"""Description----------UniProt特征分析结果类, 存储从UniProt数据库获取的蛋白质特征信息列表Args----------features (List[UniProt_Feature]): UniProt特征列表Returns----------Dict[str, List[UniProt_Feature]]: 以特征类型为键, 特征列表为值的字典, 也就是features按类型分类存储;所以同一类型的feature会被存储在同一个列表中"""features: Dict[str, List[UniProt_Feature]]def get_uniprot_features(protein_id: str) -> UniProt_Feature_Analysis_Result:"""Description----------从UniProt数据库获取指定蛋白质的特征信息Args----------protein_id (str): UniProt蛋白质IDReturns----------UniProt_Feature_Analysis_Result: UniProt特征分析结果对象"""url = f"https://rest.uniprot.org/uniprotkb/{protein_id}"# 指定返回JSON格式headers = {"Accept": "application/json"}response = requests.get(url, headers=headers, timeout=10)if response.status_code == 200:data = response.json()# 初始化features字典, feature_type作为键, 对应的UniProt_Feature列表作为值features: Dict[str, List[UniProt_Feature]] = {}for feature in data.get("features", []):f_type = feature.get("type")f_description = feature.get("description", "")f_start = feature["location"]["start"]["value"]f_end = feature["location"]["end"]["value"]uni_feature = UniProt_Feature(feature_type=f_type,start=f_start,end=f_end,description=f_description)# 如果该feature_type还未在字典中, 则初始化一个空列表if f_type not in features:features[f_type] = []# 将当前feature添加到对应类型的列表中features[f_type].append(uni_feature)return UniProt_Feature_Analysis_Result(features=features)
分析结果如下,
因为这是一个特定的dataclass分析对象,所以我们还得解包:
result = get_uniprot_features("P49711")
for ftype, flist in result.features.items():print(f"Feature Type: {ftype}")for feature in flist:print(f" Start: {feature.start}, End: {feature.end}, Description: {feature.description}")
这样其实就顺眼多了: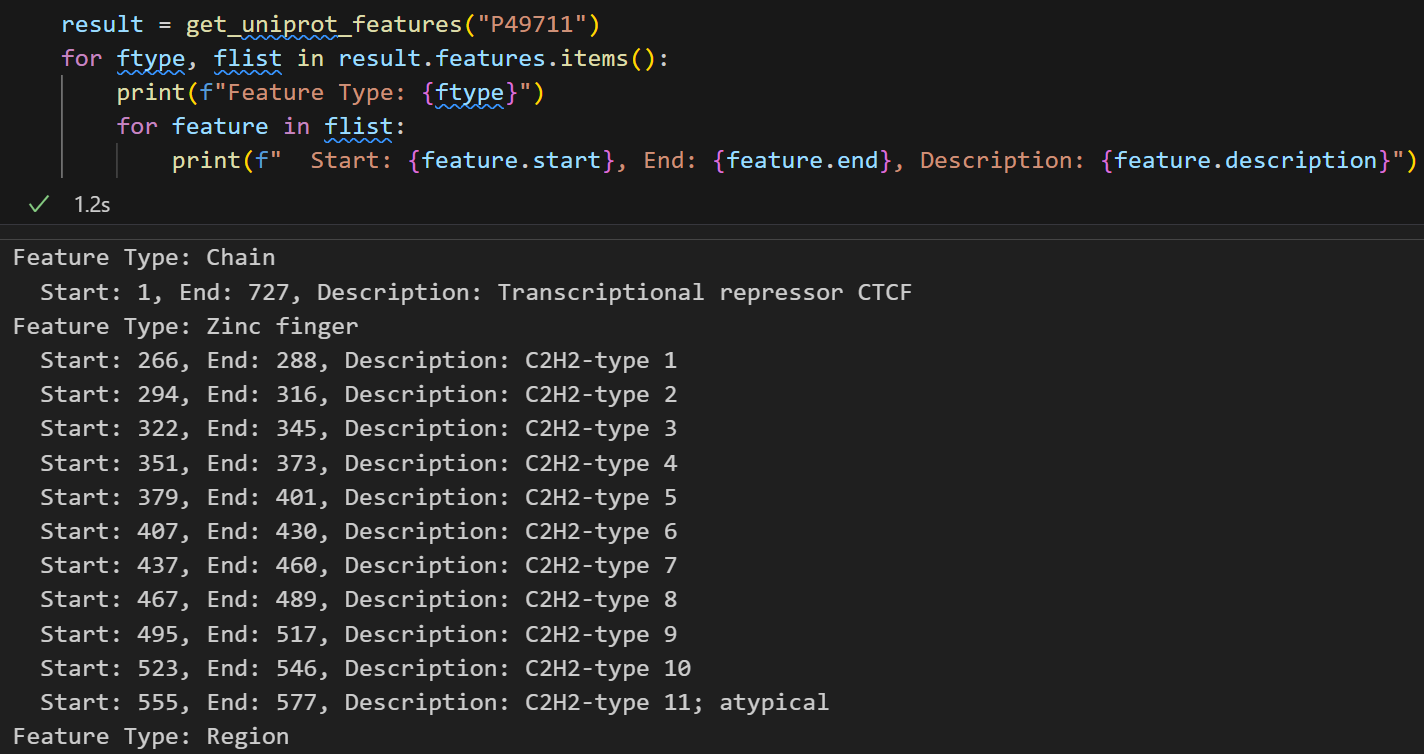
如果是结构域的话,我们可以只抓取其中domain、zinc finger之类的feature类型,稍微在上面的代码中改一改即可,比如说加一行判断,类似于:
if feature['type'] in ['DOMAIN', 'REGION', 'FAMILY']:
(3)一些补充:
除了(2)中从纯数据条目返回的json格式数据中解包,再一个一个找feature的键值对之外;
其实还有更简单的方法,就是使用api,我们其实同样可以从query中添加类似于feature直接请求的参数,可以获取feature相关的数据:
参考:https://www.uniprot.org/help/sequence_annotation

https://www.uniprot.org/help/general_annotation

其实逻辑和前面构建query的(1)一样,非常简单,所以就在这里停住,我会在下一篇博客中继续整理。