【文献分享】KADAIF:一种针对复杂微生物组数据的异常检测方法
文章目录
- 介绍
- 代码
- 参考
介绍
肠道微生物群在人类健康和疾病中起着重要作用,因此引发了大规模的研究,从而产生了大量的数据集。在分析此类数据集时,一个关键的预处理步骤是异常检测,其目的是识别错误样本并防止产生误导性的统计结果。然而,微生物组数据存在独特的挑战,如组成性、稀疏性、相互依赖性和高维度性,这限制了传统方法的有效性,并凸显了在微生物组数据中进行异常检测时需要专门定制的方法。
为应对这一挑战,我们引入了 KADAIF,这是一种针对微生物组的特定异常检测方法,它扩展了常见的孤立森林(IF)方法。与 IF 类似,KADAIF 构建了一个树的集合,每个树都通过随机选择的特征递归地对数据进行划分,并测量样本被孤立的平均深度,假设异常样本会更接近树的根部被孤立。然而,与 IF 不同的是,KADAIF 是基于特征子集(结合降维技术)对样本进行划分,从而解决了微生物组特有的特性,如稀疏性和物种相互作用。
我们通过模拟引入异常行为的常见场景来评估 KADAIF,结果表明 KADAIF 在各种设置和数据集上均优于其他方法。此外,我们还展示了 KADAIF 在检测其他类型的高维稀疏生物数据中的异常情况方面也优于 IF。最后,我们展示了 KADAIF 在纵向微生物组数据中用于识别疾病发作以及基于安娜·卡列尼娜原则对病例和对照进行划分的应用。综合来看,我们的工作突显了 KADAIF 在增强微生物组数据处理和后续分析方面的潜力,这对精准医学研究具有有益的影响。
The gut microbiome plays an important role in human health and disease, prompting large-scale studies that generate extensive datasets. A critical preprocessing step in analysing such datasets is anomaly detection, which aims to identify erroneous samples and prevent misleading statistical outcomes. Microbiome data, however, pose unique challenges such as compositionality, sparsity, interdependencies, and high dimensionality, limiting the effectiveness of conventional methods and highlighting the need for specifically-tailored approaches for anomaly detection in microbiome data.
To address this challenge, we introduce KADAIF, a microbiome-specific anomaly detection method that generalizes the common Isolation Forest (IF) approach. As in IF, KADAIF builds an ensemble of trees, each recursively partitioning the data along randomly selected features, and measures the average depth at which samples are isolated, assuming that anomalous samples will be isolated closer to the root. Unlike IF, however, KADAIF partitions samples based on subsets of features (coupled with dimensionality reduction), addressing microbiome-specific properties such as sparsity and species interactions.
We evaluate KADAIF by simulating common scenarios that introduce anomalous behavior, demonstrating that KADAIF outperforms alternative methods across various settings and datasets. Furthermore, we show that KADAIF outperforms IF in detecting anomalies also in other types of high-dimensional sparse biological data. Finally, we show KADAIF’s application for identifying disease onset in longitudinal microbiome data and for partitioning cases versus controls based on the Anna Karenina principle. Combined, our work highlights KADAIF’s potential to enhance microbiome data processing and downstream analyses, with beneficial implications for precision medicine studies.
人类微生物群,尤其是人类肠道微生物群(即存在于人体肠道内的所有微生物集合体),在各种健康状况中发挥着明确的作用(吉尔伯特等人,2018 年;范和佩德森,2021 年;沙纳汉等人,2021 年)。肠道微生物群在我们健康中的这种作用促使人们对这一复杂系统进行了大量深入研究,对不同群体和疾病状态下的微生物群组成进行了全面分析(科西奇等人,2014 年;施里纳尔等人,2015 年;江等人,2017 年;沃德罗等人,2018 年;亚奇达等人,2019 年)。这些研究反过来又产生了大量大规模的数据集,这些数据集可以进行处理和分析,以获得有关健康和疾病中微生物群的新见解(赫滕霍弗等人,2012 年;梅特等人,2012 年;泽尔纳科娃等人,2016 年;麦克唐纳等人,2018 年;普罗克特等人,2019 年;弗朗廷等人,2022 年)。
一项相关任务是在从大规模临床研究中获取的数据的预处理阶段常见的做法,即异常检测(在不同领域也被称为离群点检测)(Fitriyani 等人,2019 年;Ripan 等人,2021 年;Shaikh 等人,2023 年)。异常指的是在数据集中的大多数样本的特征方面存在某种偏离的数据样本,这可能表明各种问题,如测量误差、错误标注、污染或批次效应。如果忽略这些异常样本,可能会引入虚假信号或掩盖真实的生物学模式,最终导致误导或令人困惑的结果。因此,检测这些异常样本并在后续分析中对其进行处理(例如从数据集中删除它们、纠正异常值、减少其在预测模型中的权重或应用更抗异常的分析方法)至关重要。
考虑到这一点,此前已经介绍了多种异常检测算法。这些算法通常采用无监督或半监督的方法,并且可以大致分为几个不同的类别(钱多拉等人,2009 年)。它们包括:(i)基于分类的技术,利用分类模型来识别异常;(ii)基于最近邻的技术,通过检查样本的接近程度来识别离群值;(iii)基于聚类的技术,将相似的样本分组并检测与已识别聚类的偏差;(iv)统计技术,通过建模数据行为来检测异常数据点;以及(v)谱技术,利用降维来识别不规则样本。
在微生物组领域,许多研究不幸地完全忽略了在进行后续分析之前检测异常情况这一任务,部分原因是缺乏适用于微生物组数据的成熟且标准的异常检测协议。在相对较少的尝试明确纳入异常检测的微生物组研究中(贝洛基奇等人,2019 年;利厄等人,2021 年;利尼亚雷斯-布兰科等人,2022 年;班贾克等人,2023 年),大多数都采用了通用方法,其中最常用的可能是孤立森林(IF)(刘等人,2008 年)。这种流行的无监督非参数异常检测方法基于构建多个随机隔离树,每个树都通过沿随机选择的特征使用随机分割点对数据空间进行递归分区。通常被认为具有独特性、罕见性和非典型特征值的异常值,预计会在这些树上被隔离,其划分的层次较少(因此更接近树的根部),而与正常样本相比更为集中。在这一由随机隔离树构成的森林中,样本被分离时的平均深度(即从根部算起的距离)可以作为异常得分的依据,平均深度越低则表明样本越异常。此外,这一概念还衍生出了几种变体(如 Sun 等人 2019 年、Li 等人 2020 年、Chabchoub 等人 2022 年所提出的),例如扩展隔离森林(Hariri 等人 2021 年),它在每个分支节点处使用随机斜率和截距,从而能够实现非轴平行的分割,并实现更灵活和细致的样本划分。
然而,值得注意的是,微生物组数据具有若干独特的特性,包括特定的组成性(格洛尔等人,2017 年)、稀疏性(齐利米格拉斯和福多尔,2016 年;潘,2021 年)以及高维度性(李,2015 年;齐利米格拉斯和福多尔,2016 年),因此通常需要专门设计的分析方法来进行整理、处理和分析。微生物组数据的这些独特特征对于异常检测尤其具有挑战性,这可能是此类方法在该领域应用有限的原因。例如,在高维度和稀疏的空间中,距离和邻域变得不那么具有信息价值(贝耶等,1999 年),从而阻碍了传统基于邻居或基于聚类的技术在检测异常方面的有效性。同样,由于微生物组
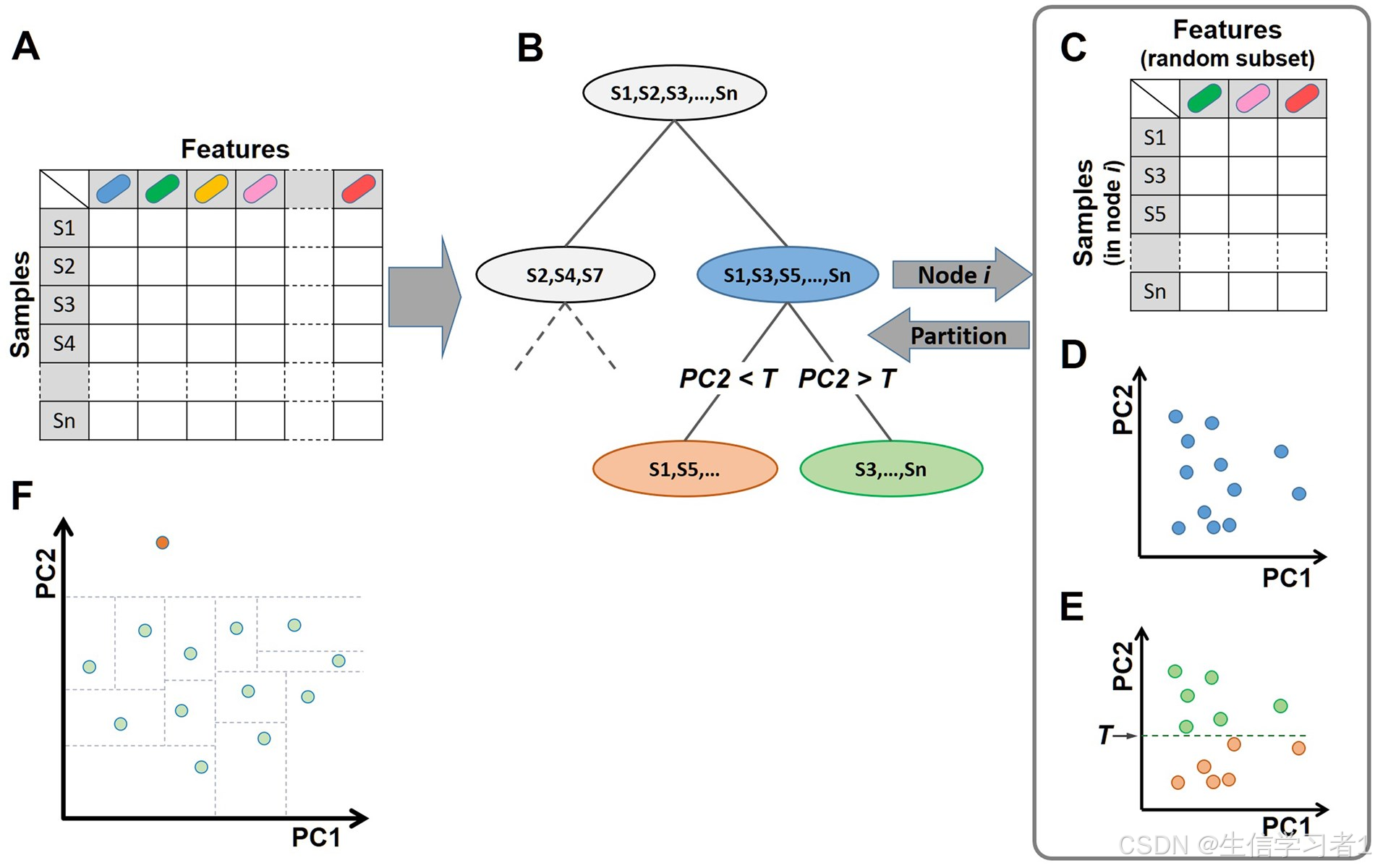
KADAIF 的示意图。(A)KADAIF 接收一个特征表作为输入,对于微生物组数据而言,该表列出了每个样本中每个分类群的丰度。(B)然后,KADAIF 开始构建一个随机隔离树的森林,其中每个节点根据某些特征将样本划分成两个子节点。具体而言,KADAIF 首先选择一个随机特征子集(C),对该子集应用降维方法(例如 PCoA)(D),并使用随机选择的某个成分上的投影来根据随机选择的阈值 T 对样本进行划分(E)。(F)与 IF 一样,KADAIF 假定异常值是独特的、罕见的且不同的,因此它们在隔离树中往往通过较少的随机分裂(用虚线表示)来孤立。在这个示例中,最上面的样本代表一个异常值,确实可以通过一次随机分裂轻松将其孤立(请注意,该样本与其余样本之间在 PC2 方面存在较大的差距,这使得可以使用广泛的随机阈值来对其进行孤立)。相比之下,大多数正常样本需要多次高度特定的分裂才能在随机情况下实现完全孤立。
代码
https://github.com/borenstein-lab/KADAIF
参考
- KADAIF: an anomaly detection method for complex microbiome data
- https://github.com/borenstein-lab/KADAIF