使用hhblits进行序列比对
一、安装hhsuit
二、查看是否安装了hhblits
Linux的查找方法:
-
打开终端:打开一个终端窗口
-
输入以下命令
hhblits --version以及
which hhblits第一个命令将显示HHblits的版本信息,而第二个命令将显示HHblits可执行文件的路径。
-
检查输出:如果看到HHblits的版本信息或可执行文件的路径,则表示HHblits已成功安装在您的系统上。如果没有任何输出或者显示"command not found",则说明HHblits尚未安装或没有正确安装。
三、准备输入文件
将待比对的蛋白质序列保存为一个FASTA格式的文件(例如,input.fasta),确保每个序列有一个唯一的标识符。
如何找一个fasta文件(可略)
1.进入ASD数据库 ASD数据库地址
2.找到MOLICULES->PROTEIN(即蛋白质)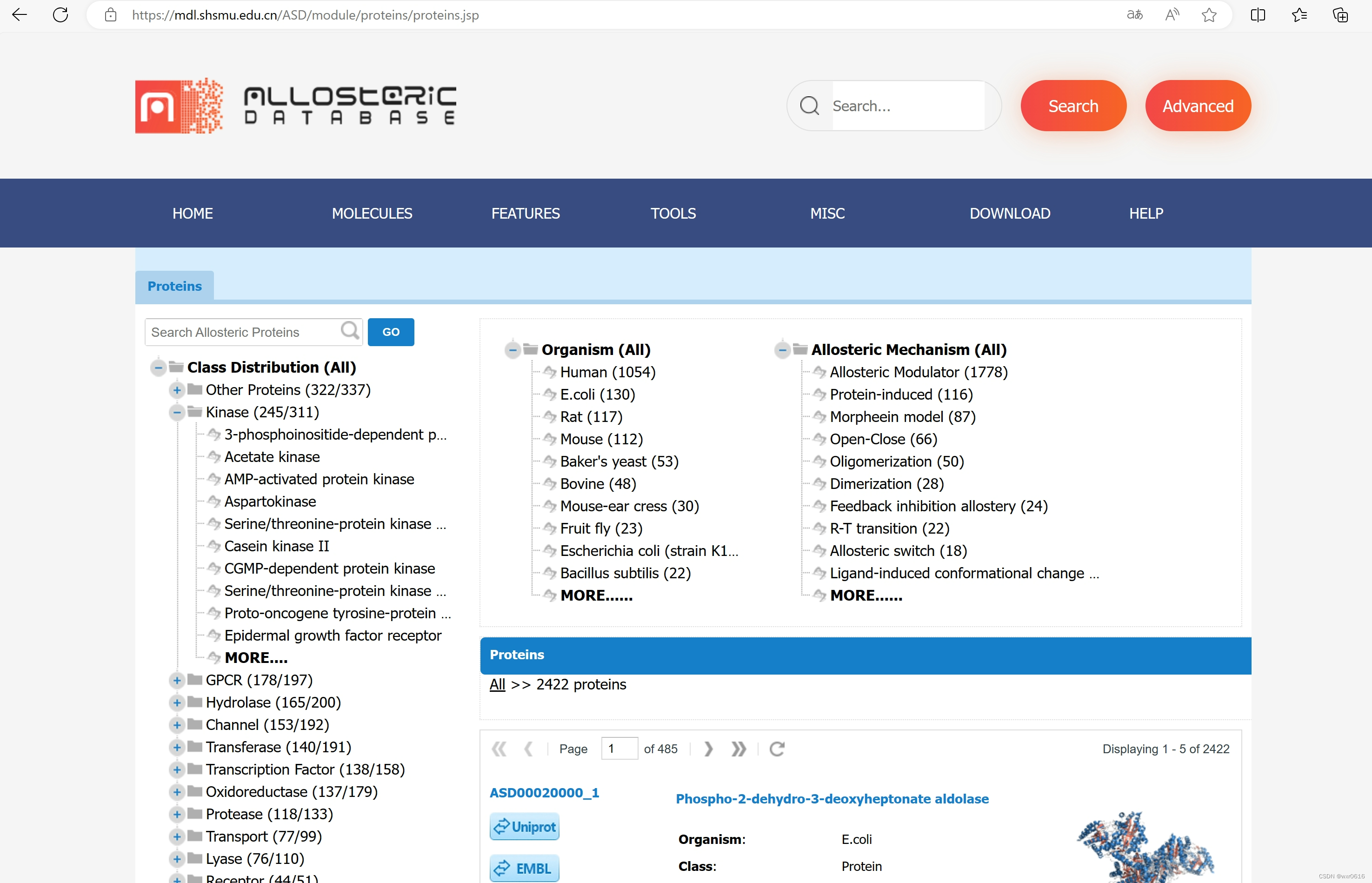
选中一个蛋白质
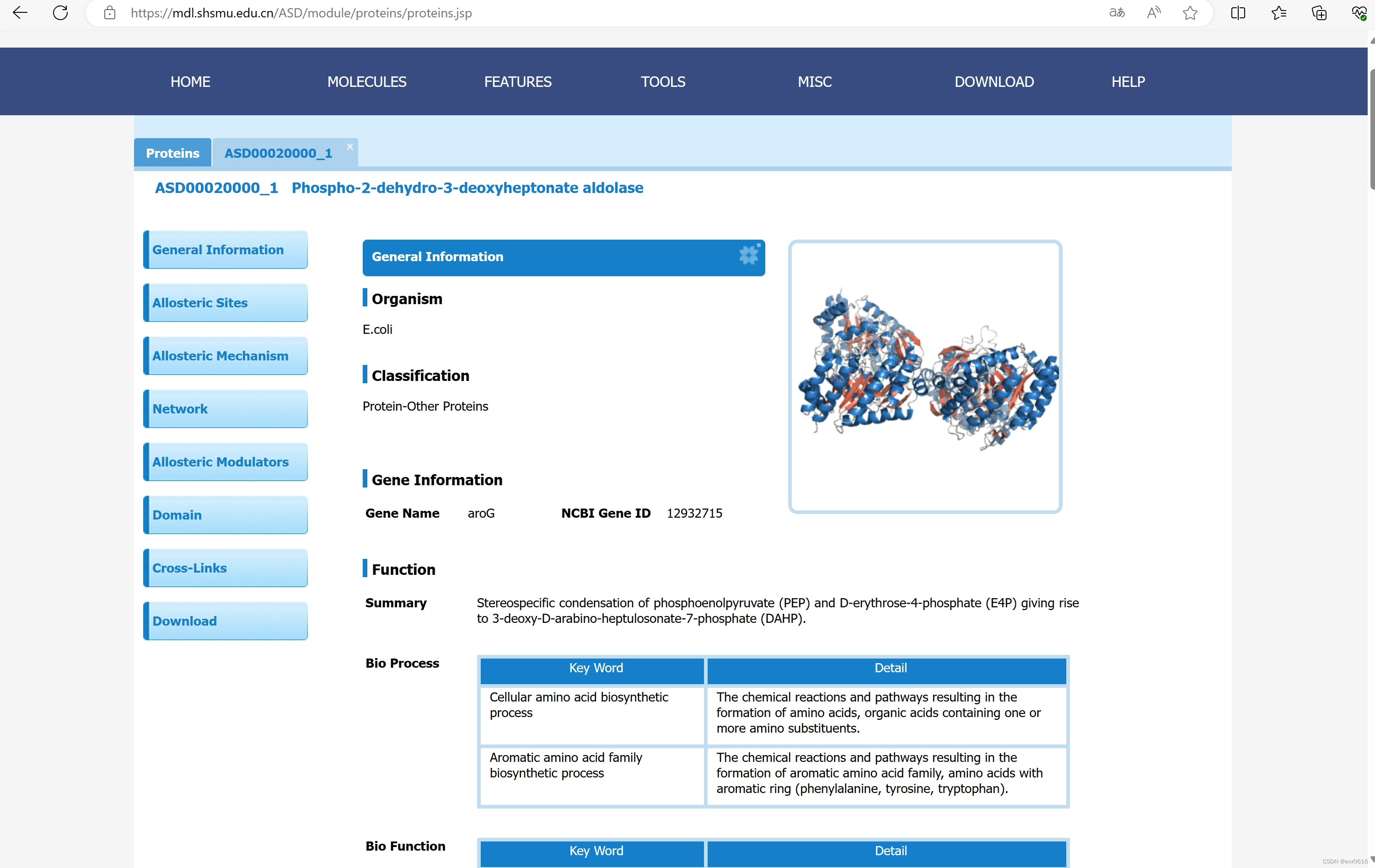
点击左侧的download,下载xml文件,打开后找到<PDB_ID>,会有很多个,代表不同序列在pdb数据库中的id,选择其中一个<PDB_ID>,例如<PDB_ID>3QCY</PDB_ID>我选择3QCY
在PDB数据库中搜索 3QCYPDB数据库地址![]() https://www.rcsb.org/
https://www.rcsb.org/
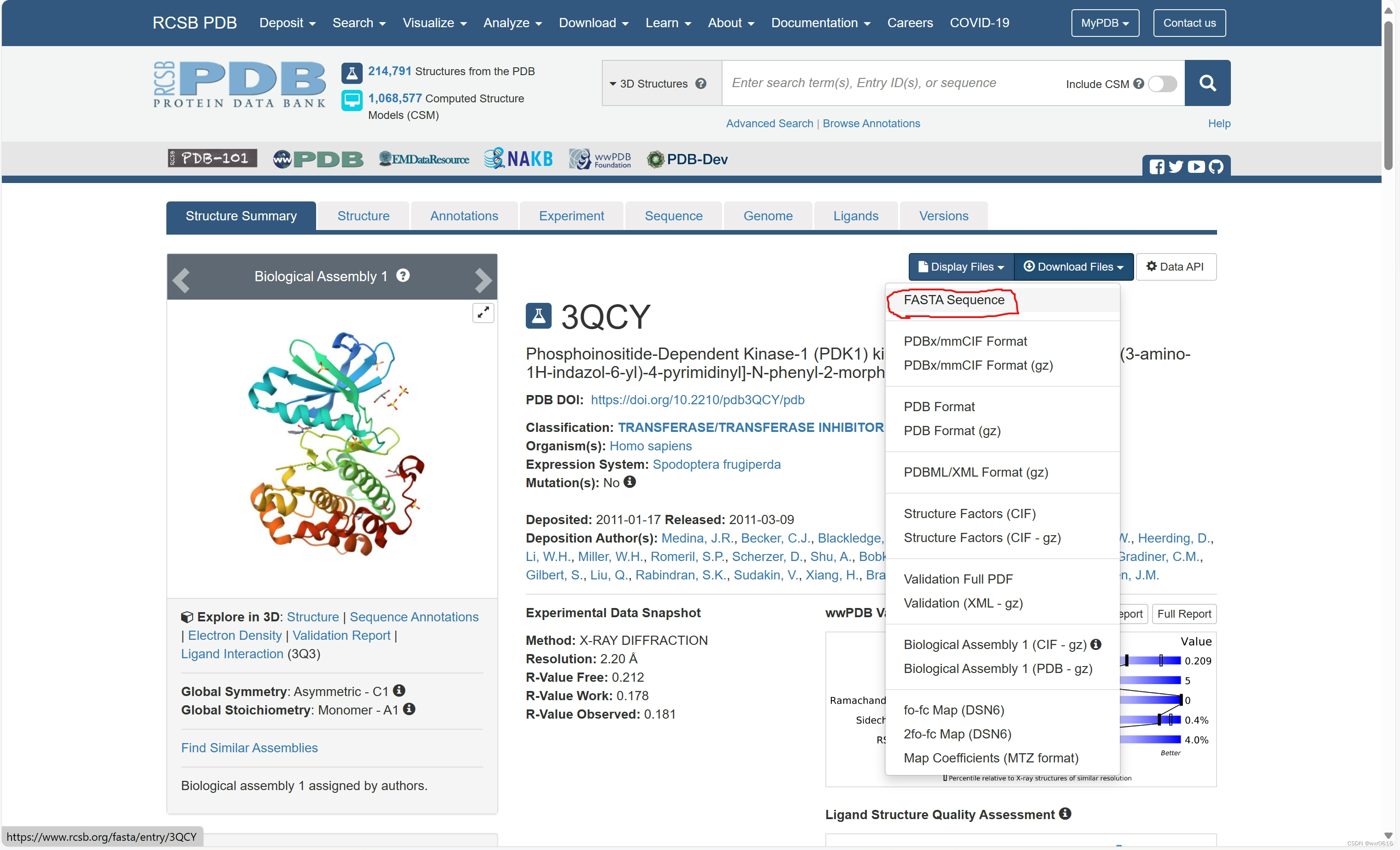
点击搜索并选择download files下载对应序列的fasta文件(如果只查看的话选择display files的fasta文件即可)
点开下载的fasta文件就可以查看<PDB_ID>为3QCY的序列了
四、构建数据库:HHblits使用预先构建的数据库来进行比对。
如何构建一个自己的PDB数据库(Linux版)
-
获取PDB数据:在终端中使用wget或curl等命令下载所需的PDB文件。例如:
wget https://ftp.rcsb.org/pub/pdb/data/structures/all/pdb/pdbXXXX.pdb.gz这里,
pdbXXXX是PDB文件的标识符。 -
解压缩PDB文件:使用gzip或gunzip命令解压缩下载的PDB文件。例如:
gunzip pdbXXXX.pdb.gz这将生成解压后的PDB文件(pdbXXXX.pdb)。
-
提取序列信息:使用pdbseq或pdb2fasta工具从PDB文件中提取蛋白质的序列信息。这些工具通常随着HH-suite软件包一起安装。示例命令如下:
pdbseq pdbXXXX.pdb > output.fasta这里,
pdbXXXX.pdb是解压后的PDB文件,output.fasta是生成的FASTA格式序列文件。 -
格式化数据库文件:使用hhmake命令将FASTA格式的序列文件转换为HHblits数据库文件(.hmm)。示例命令如下:
hhmake -i output.fasta -o database.hmm这里,
output.fasta是上一步生成的FASTA格式序列文件,database.hmm是生成的HHblits数据库文件。 -
构建索引:使用hhindex命令对数据库文件进行索引以提高比对效率。示例命令如下:
hhindex -i database.hmm -o index.hhm这里,
database.hmm是上一步生成的HHblits数据库文件,index.hhm是生成的索引文件。 -
设置环境变量:将数据库和索引文件的路径添加到环境变量中,以便HHblits可以找到它们。您可以在.bashrc或.bash_profile等配置文件中添加以下行:
export HHBLITS_DB=path_to_database export HHBLITS_INDEX=path_to_index这里,
path_to_database是您数据库文件的所在路径,path_to_index是索引文件的所在路径。
由于我已经下载好了对应的fasta文件,所以直接转成hmm文件即可
hhmake -i /home/fasta/rcsb_pdb_3QCY.fasta -o rcsb_pdb_3QCY.hmm