生信分析自学攻略 | R语言函数与参数介绍
在《生信小白自学攻略》系列的前几篇文章中,我们已经掌握了 R 语言的数据类型、数据结构以及基础的数据处理方法。然而,如果你发现自己在重复编写相似的代码,或者想要将一段逻辑封装起来方便复用,那么恭喜你,是时候深入了解 R 语言的函数了!它是R语言中组织代码、实现自动化、提高效率的关键。本篇将深入讲解函数的定义、参数应用,并通过实例助你构建专属分析工具箱,让代码更高效、简洁、易维护。
函数:代码的“积木”
什么是函数?
函数是R语言中一段被命名、可重复使用的代码块。它接收输入(参数),执行特定任务,并返回结果。你可以将其视为一个“黑箱”:你给它输入,它就给你输出。R内置了许多常用函数,如 mean()、sum() 等。而现在,我们将学习如何创建自己的“积木”。
为什么需要函数?
代码复用: 避免重复编写相同的代码,提高效率。
模块化: 将复杂问题分解为小模块,提高代码可读性和可维护性。
可读性: 函数名称本身能说明其功能,使代码逻辑更清晰。
易于调试: 当出现问题时,能更快定位到特定函数。
例如,你需要对多个基因表达矩阵进行归一化。没有函数,你将为每个矩阵重复编写代码;有了函数,只需一行代码调用即可完成。
函数的定义与调用
函数的定义
在R中,使用 function()用 function() 关键字来定义一个函数。基本语法如下:
function_name <- function(parameter1, parameter2, ...) {# 函数体:包含执行任务的代码return(result) # 可选:返回函数执行的结果
}
function_name: 你为函数取的名称,应清晰有意义。function(): 定义函数的关键字。parameter1,parameter2, ...: 函数的参数列表,接收输入值。参数是可选的。{}: 函数体,包含函数要执行的所有代码。return(result): 显式返回函数执行结果。若无return(),函数将默认返回函数体中最后一行表达式的值。
示例:一个简单的加法函数
# 定义一个名为 add_numbers 的函数,它接受两个参数 x 和 y
add_numbers <- function(x, y) {sum_val <- x + yreturn(sum_val) # 返回它们的和
}
函数的调用
定义函数后,通过其名称并提供实际参数来调用它。
# 调用 add_numbers 函数
result1 <- add_numbers(5, 3)
print(result1) # 输出: 8

函数参数的深度探索
参数是函数与外部世界交互的桥梁。理解不同类型的参数及其行为,对于编写灵活且健壮的函数至关重要。
形式参数与实际参数
形式参数: 定义函数时在括号内声明的变量名,如 add_numbers 中的 x 和 y。
实际参数: 调用函数时传递给函数的值,如 add_numbers(5, 3) 中的 5 和 3。
参数匹配方式
位置匹配
实际参数按照顺序与形式参数一一对应。
subtract_numbers <- function(a, b) {return(a - b)
}# 位置匹配:a 对应 10,b 对应 4
result <- subtract_numbers(10, 4)
print(result) # 输出: 6
名称匹配
通过参数名称来指定实际参数,无需考虑顺序,提高了代码可读性。
# 再次使用 subtract_numbers 函数
result_named <- subtract_numbers(b = 4, a = 10) # 顺序无关紧要
print(result_named) # 输出: 6

最佳实践: 推荐在函数调用中使用名称匹配,尤其当函数参数较多时。
混合匹配
可混合使用位置匹配和名称匹配。一旦开始使用名称匹配,后续参数也必须使用名称匹配。
mixed_result <- subtract_numbers(10, b = 4) # 10通过位置匹配给a,4通过名称匹配给b
print(mixed_result) # 输出: 6
默认参数值
定义函数时可为参数指定默认值。若调用时未提供该参数的值,则使用默认值,使函数更灵活。
# 计算平均值的函数,可以指定是否移除 NA 值
calculate_mean <- function(data_vector, na.rm = TRUE) {
if (na.rm) {data_vector <- na.omit(data_vector)}
return(mean(data_vector, na.rm = FALSE)) # na.rm=FALSE 是为了避免mean函数内部再次处理NA
}values <- c(1, 2, 3, NA, 5)# 使用默认值 na.rm = TRUE
mean1 <- calculate_mean(values)
print(mean1) # 输出: 2.75# 明确指定 na.rm = FALSE
mean2 <- calculate_mean(values, na.rm = FALSE)
print(mean2) # 输出: NA

变长参数 ... (Dot-Dot-Dot Argument)
... 允许函数接受任意数量的额外参数,并将这些参数传递给函数内部的其他函数。这在编写通用函数时非常有用。
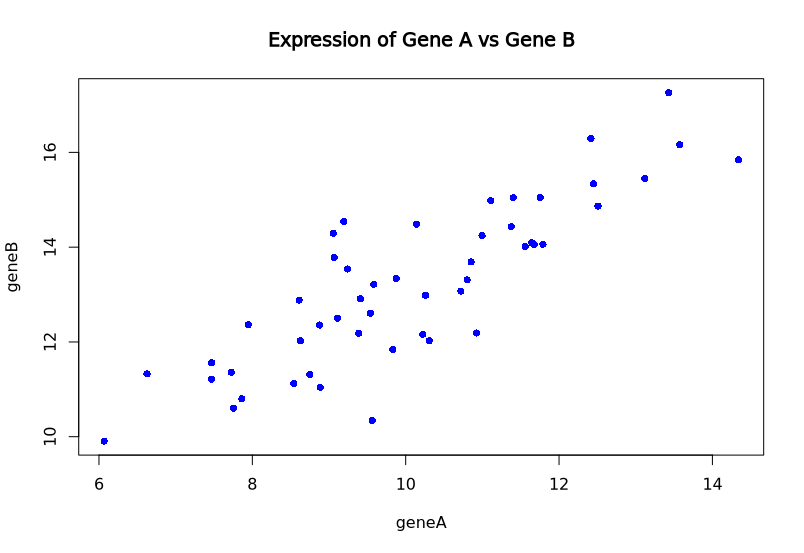
示例:绘制散点图的包装函数
# 绘制基因表达散点图的函数
plot_gene_expression <- function(gene1_expression, gene2_expression, title = "Gene Expression Scatter Plot", ...) {plot(gene1_expression, gene2_expression,main = title,xlab = deparse(substitute(gene1_expression)),ylab = deparse(substitute(gene2_expression)),...) # 将所有额外的参数传递给 plot 函数
}# 创建示例数据
set.seed(123)
geneA <- rnorm(50, mean = 10, sd = 2)
geneB <- geneA * 0.8 + rnorm(50, mean = 5, sd = 1)# 调用函数,并传递额外的图形参数
plot_gene_expression(geneA, geneB,title = "Expression of Gene A vs Gene B",col = "blue", # 传递给 plot() 的颜色参数pch = 16) # 传递给 plot() 的点类型参数
在这个例子中,plot_gene_expression 内部调用了 plot() 函数,... 参数使得我们可以将 col、pch 等 plot() 特有的参数直接传递进去。
生物信息学案例:计算基因表达量统计信息
实操步骤:计算基因表达量均值、中位数和标准差
编写一个函数,接收一个基因表达量的数值向量,计算其均值、中位数和标准差,并返回一个命名向量。此函数在初步探索基因表达数据分布时非常有用。
#' 计算基因表达量的统计信息
#'
#' @param expression_values 一个数值向量,代表基因表达量。
#' @param na_action 字符串,指定如何处理缺失值 (NA)。
#' "remove" (默认) 移除 NA;"keep" 保留 NA;"fail" 如果有 NA 则报错。
#' @return 一个命名数值向量,包含均值 (mean)、中位数 (median) 和标准差 (sd)。
#' @examples
#' gene_exp <- c(10.5, 12.1, 8.9, 15.0, NA, 11.2)
#' calculate_expression_stats(gene_exp)
#' calculate_expression_stats(gene_exp, na_action = "keep")
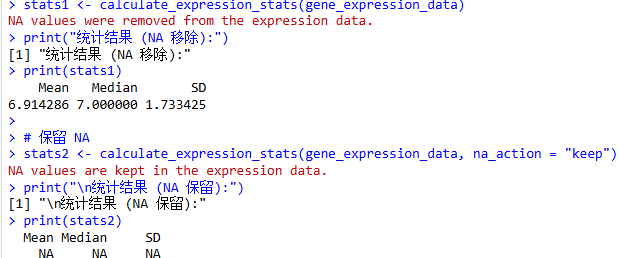
calculate_expression_stats <- function(expression_values, na_action = "remove") {
if (!is.numeric(expression_values)) {stop("Input 'expression_values' must be a numeric vector.")}if (any(is.na(expression_values))) {if (na_action == "remove") {expression_values <- na.omit(expression_values)message("NA values were removed from the expression data.")} elseif (na_action == "fail") {stop("NA values found in expression data. Set 'na_action' to 'remove' or 'keep' to proceed.")} elseif (na_action == "keep") {message("NA values are kept in the expression data.")} else {stop("Invalid 'na_action' specified. Choose 'remove', 'keep', or 'fail'.")}}mean_val <- mean(expression_values, na.rm = FALSE)median_val <- median(expression_values, na.rm = FALSE)sd_val <- sd(expression_values, na.rm = FALSE)stats <- c(Mean = mean_val, Median = median_val, SD = sd_val)
return(stats)
}# 示例使用
gene_expression_data <- c(5.2, 7.8, 6.5, 9.1, 4.3, 7.0, NA, 8.5)# 默认行为:移除 NA
stats1 <- calculate_expression_stats(gene_expression_data)
print("统计结果 (NA 移除):")
print(stats1)# 保留 NA
stats2 <- calculate_expression_stats(gene_expression_data, na_action = "keep")
print("\n统计结果 (NA 保留):")
print(stats2)
通过此函数,你可以方便地对任何基因表达量向量进行快速统计,无论是单个基因在不同样本的表达,还是某个样本中所有基因的表达分布。
小结
本篇教程深入讲解了R语言中函数的核心概念。我们学习了如何定义和调用函数,并详细探讨了函数参数的各种类型,包括位置匹配、名称匹配、默认参数以及强大的 ... 变长参数。通过一个贴近生物信息学实际的案例,你已经掌握了如何编写和应用自定义函数来计算基因表达量的统计信息。
掌握函数是R语言进阶的关键一步。它能帮助你将重复性任务自动化,提高代码的可读性、可维护性和复用性,从而更高效地进行生物信息学数据分析。