scanpy单细胞转录组python教程(一):不同形式数据读取
过去我们主要的单细胞分析使用的是R语言seurat进行,主要考虑的是大多数人的情况,很多人没有接触过python。但是,随着很多个性化分析的需求,以及细胞量级的增加,python分析单细胞也是一个必须要掌握的内容了,与R对接的python版是scanpy,它是一个与 anndata 联合开发的用于分析单细胞基因表达数据的可扩展工具包。它包含单细胞数据预处理、可视化、聚类、轨迹推断和差异表达测试等功能。基于 Python 的实现能够高效处理超过一百万个细胞的数据集,这一点也是它最大的优势,随着单细胞数据量的增加,seurat处理内存明显吃紧,运行速度可能会慢,而且大型数据集没法处理,所以scanpy成了不得不学习的对象。当然了,R和python各有优势,个人经验而言,R的可视化更好更方便,但是目前有很多途径能够实现seurat object和scanpy的转化,所以功能互用不是问题。从这里开始我们正式介绍scanpy分析流程,就类似于R中seurat前期的处理一样,从数据读取、降维聚类、细胞注释到基础可视化就完成了一个完整的流程,最终得到了我们整个数据分析最重要的基石-注释好的单细胞数据,然后就可以用于个性化分析及数据探索了!官网教程:
https://scanpy.readthedocs.io/en/stable/index.html
anndata数据结构:
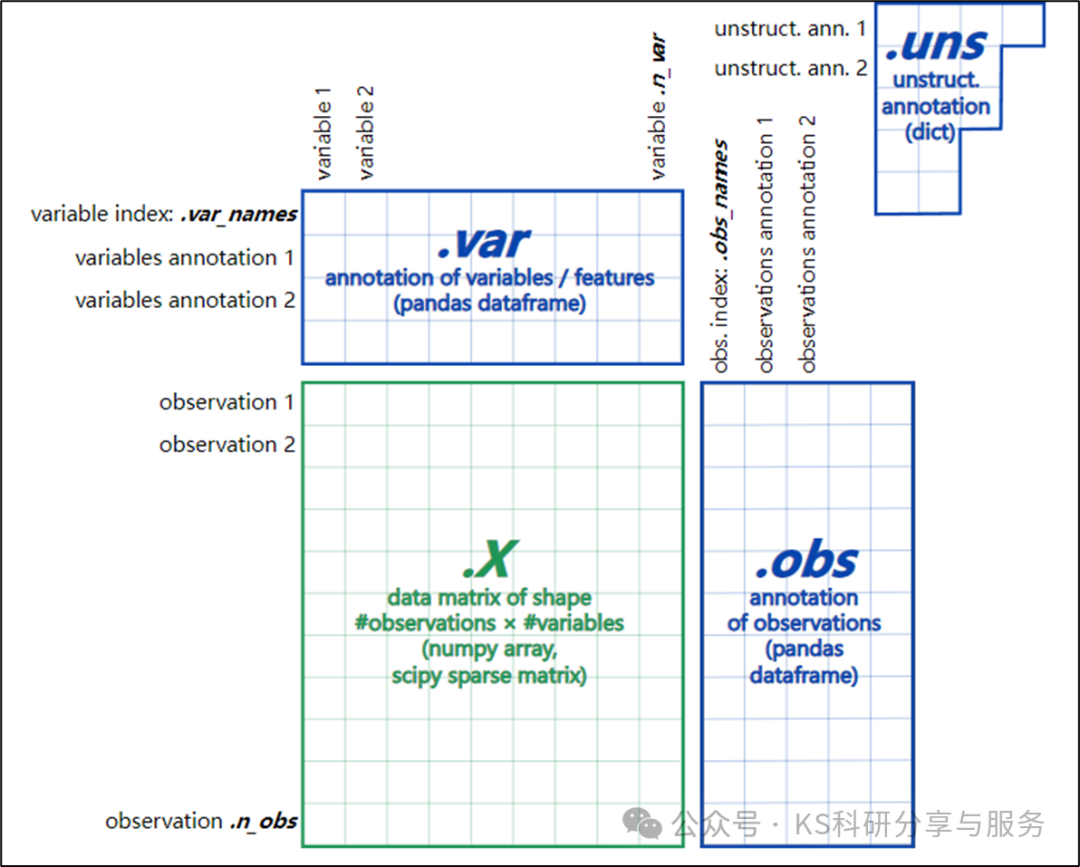
在分析前,最好单独创建一个环境:
#分析最好还是建立单独环境
conda create -n scanpy python=3.10
conda activate scanpy
#安装
pip install scanpy -i https://pypi.tuna.tsinghua.edu.cn/simple
#Successfully installed anndata-0.11.4 array-api-compat-1.11.2 contourpy-1.3.2 cycler-0.12.1 fonttools-4.58.0 h5py-3.13.0
#joblib-1.5.0 kiwisolver-1.4.8 legacy-api-wrap-1.4.1 llvmlite-0.44.0 matplotlib-3.10.3 natsort-8.4.0 networkx-3.4.2
#numba-0.61.2 numpy-2.2.5 pandas-2.2.3 patsy-1.0.1 pillow-11.2.1 pynndescent-0.5.13 pyparsing-3.2.3 pytz-2025.2
#scanpy-1.11.1 scikit-learn-1.5.2 scipy-1.15.3 seaborn-0.13.2 session-info2-0.1.2 statsmodels-0.14.4 threadpoolctl-3.6.0
#tqdm-4.67.1 tzdata-2025.2 umap-learn-0.5.7接下来以单样本为例,演示一下scanpy到底有什么流程,熟悉它的操作。单细胞数据读取:基本上scanpy提供了所有格式的数据读取函数,例如以下等等sc.read_10x_h5()、
sc.read_10x_mtx、sc.read_csv、sc.read_h5ad、sc.read_loom。看这些函数的名称就知道他们是读取什么格式的函数了。所以我们可以仅用sc的函数就可以完成各类型数据的读取了!其实无论什么格式的数据,读取的都是表达矩阵。
#导入库
import scanpy as sc
import pandas as pd
import numpy as np
sc.settings.verbosity = 3 # verbosity: errors (0), warnings (1), info (2), hints (3) #控制 Scanpy 运行时输出的日志详细程度
sc.logging.print_header()
sc.settings.set_figure_params(dpi=300, facecolor="white")第一种数据格式:10X经典三个文件mtx
adata = sc.read_10x_mtx("./单样本数据/10x_mtx/", # the directory with the `.mtx` file,文件目录var_names="gene_symbols", # use gene symbols for the variable names (variables-axis index)cache=True, # write a cache file for faster subsequent reading
)
adata
AnnData object with n_obs × n_vars = 4968 × 33538var: 'gene_ids', 'feature_types'第二种数据格式:h5文件:跑过上游的小伙伴应该知道,输出的结果文件,https://mp.weixin.qq.com/s/X7pe5aOoiIyncAc5cjgxBA,我这里以10X ATAC的结果为例看看了,RNA的没有了,其实都差不多。我们可以发现,除了经典的三个文件,还有一个h5文件,就是专门针对python的,这个文件也是表达矩阵,可以直接读取。
#一个h5的数据,是2025年的,因为是压缩文件,我们先解压一下:
! tar -xvf ./单样本数据/h5/GSE273145_RAW.tar
adata = sc.read_10x_h5('./GSM8422600_p5s2.h5')
adata第三种数据格式:txt或者csv文件:这种通常是直接提供了txt或者csv格式的表达矩阵!都是使用sc.read_csv。读取csv文件的时候需要注意,你需要事先确认矩阵的形式,因为sc.read_csv() 默认将行作为观测(obs-cellid),列作为变量(var-gene)。所以正常的格式要求是行为细胞,列为基因!!!。如果你的矩阵行为基因,列为细胞,就需要转置一下。我们此处演示的数据就是如此,所以读取的时候加.T。
!gunzip ./单样本数据/file_txt/GSM5098737_HM1.csv.gz #解压
adata1 = sc.read_csv('./单样本数据/file_txt/GSM5098737_HM1.csv').T对于csv或者txt文件格式的表达矩阵,有很多的读取方式。这里我们再演示一种比较常规的方法,pandas结合anndata。
df = pd.read_csv("./单样本数据/file_txt/GSM5098737_HM1.csv", index_col=0) # 第一列作为行名(基因名)
df# 转换为 AnnData
adata = sc.AnnData(df.T) # 添加基因名和细胞名
adata.var_names = df.index.values # 基因名
adata.obs_names = df.columns.values # 细胞名adata
AnnData object with n_obs × n_vars = 7228 × 17279第四种数据格式:loom文件:我这里示例的数据是一个昨晚RNA速率上游的数据,仅演示sc.read_loom读取数据。 读取loom文件需要在环境中先安装loompy: pip install loompy -i https://pypi.tuna.tsinghua.edu.cn/simple
#先解压文件
!tar -xvf ./单样本数据/loom_file/GSE226010_RAW.tar
#双重压缩,再解压一次
!gunzip ./GSM8986458_APC-T17D_ana.loom.gz
adata = sc.read_loom('./GSM8986458_APC-T17D_ana.loom')
adata
AnnData object with n_obs × n_vars = 13208 × 32285obs: 'Clusters', '_X', '_Y'var: 'Accession', 'Chromosome', 'End', 'Start', 'Strand'layers: 'matrix', 'ambiguous', 'spliced', 'unspliced'第四种数据格式:h5ad文件:这种格式也挺常见,很多公共数据库提的数据都是如此!我示例的这个数据是一个整合的数据,包括多个组!
adata = sc.read_h5ad('./单样本数据/h5ad/GSE278332_raw_count_matrix.h5ad')
adata
AnnData object with n_obs × n_vars = 16908 × 53700obs: 'time', 'group', 'condition', 'batch'var: 'gene_id'不管怎么变化,其实目的都是一样的,都是读取了基因X细胞表达矩阵,然后创建anndata,就类似于seurat中创建seuratobj一样,读入数据后进行下一步分析!