SaProt 模型部署与运行教程
写在前面:
蛋白质是生物学功能的基础,了解它们为医学、制药和基因研究开辟了有希望的途径。蛋白质语言模型(PLM)从 NLP 方法学中汲取灵感,通过对大量蛋白质 1D 残基序列进行自监督训练,被证明能够非常熟练地捕获长程残基的相关性,成为蛋白质表示的关键技术。一些著名的 PLM,如 UniRep、ProtTrans、ESM和 Evoformer在与蛋白质结构和功能相关的各种任务中展示了出色的性能。尽管基于残基序列的预训练取得成功,但鉴于蛋白质3D结构与其功能具有直接相关性,并且AlphaFold2(AF2)在蛋白质结构预测方面取得的突破已经产生了大规模的结构数据库,激发了人们对利用大规模蛋白质结构用于训练PLM的兴趣。在本文中,作者的目标是通过引入一个经过蛋白质序列和结构数据训练的大型且更强大的PLM,为生物界做出贡献。因此一个值得关注的问题在于如何将结构数据更好地引入语言模型进行预训练?
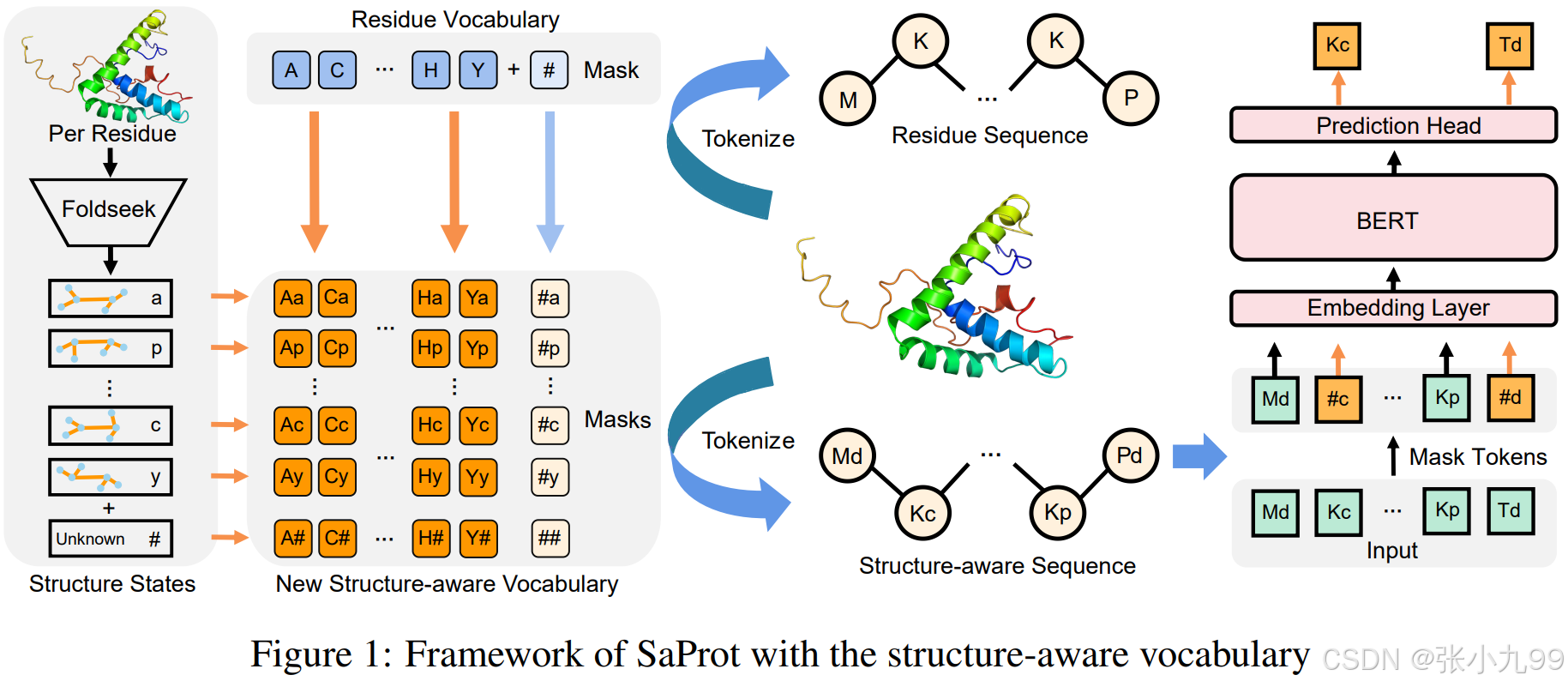
Foldseek的最初目标是促进快速准确地搜索结构相似的蛋白质,其通过将3D结构编码为3Di字母表中的序列,从而将结构比对简化为序列比对。3Di 字母表描述了3D 空间中残基 与其最近邻残基 之间的三维接触形状。从每个残基 的周围局部主链和最邻近残基 周围局部主链的6个 坐标中提取7个角度、欧几里得 距离和两个序列距离特征,这 10 个特征用于通过训练 VQ-VAE 来定义 20 个 3Di 状态,以学习最大进化守恒状态。对于给定3D结构的蛋白质,在进行结构搜索时,encoder可以预测每个残基 i 的3Di状态,将3D坐标简化为包含结构信息的序列tokens。
作者受Foldseek的启发,从新的角度整合蛋白质1D序列和3D结构,创建了结构感知(SA)词汇表,其中每个 SA tokens 都包含残基信息和结构信息。这使得原始残基序列能够转换为 SA tokens 序列,作为现有基于残基的 PLM 的输入。通过对大量蛋白质 SA-token 序列进行无监督训练,作者获得了一个名为 SaProt 的结构感知蛋白质语言模型。
一、环境安装
1、克隆模型代码和权重
# 克隆代码仓库
git clone https://github.com/westlake-repl/SaProt.git
cd SaProt# 克隆模型权重(需要 Git LFS)这里下载的是1.3B的模型,其他模型也一样
cd model
git clone https://huggingface.co/westlake-repl/SaProt_1.3B_AFDB_OMG_NCBI
cd SaProt_1.3B_AFDB_OMG_NCBI# 下载大文件(模型参数权重)
git lfs pull2、创建并激活 Conda 虚拟环境(Python 3.10)
conda create -n SaProt python=3.10 -y
conda activate SaProt
3、编辑或创建 environment.sh 文件,内容如下:
#!/bin/bash# 安装特定版本的 PyTorch(CUDA 11.7 支持)及其官方组件
pip install torch==1.13.1+cu117 torchvision==0.14.1+cu117 torchaudio==0.13.1 \--extra-index-url https://download.pytorch.org/whl/cu117# 常用工具和依赖库
pip install wandb==0.12.10 # 实验追踪工具
pip install transformers==4.28.0 # HuggingFace 模型库
pip install easydict==1.10 # 字典类配置工具
pip install biopython==1.81 # 生物信息处理库
pip install scipy==1.14.1 # 科学计算库
pip install peft==0.10.0 --no-deps # 参数高效微调,避免升级 torch
pip install lmdb==1.4.1 # 高性能嵌入式数据库
pip install torchmetrics==0.9.3 # PyTorch 指标评估库
pip install pandas==2.1.1 # 数据处理库
pip install fair-esm==2.0.0 # Meta 提供的蛋白语言模型
4、运行安装脚本
bash environment.sh5、安装兼容版本的 pytorch-lightning
由于 pip 新版本对旧 metadata 解析报错,我们需先降级 pip
pip install "pip<24.1"
pip install pytorch-lightning==1.8.3
6、补充依赖安装
pip install numpy==1.26.4
pip install protobuf==3.20.3
二、测试脚本运行
1、加载模型方式
(1)使用 Hugging Face Transformers 加载模型
请注意,遮盖 AF2 结构的较低 pLDDT 区域是有益的,详见下文。
from transformers import EsmTokenizer, EsmForMaskedLM# 加载模型
model_path = "/your/path/to/SaProt_650M_AF2" # Note this is the directory path of SaProt, not the ".pt" file
tokenizer = EsmTokenizer.from_pretrained(model_path)
model = EsmForMaskedLM.from_pretrained(model_path)# 使用模型进行推理
#################### Example ####################
device = "cuda"
model.to(device)seq = "M#EvVpQpL#VyQdYaKv" # Here "#" represents lower plDDT regions (plddt < 70)
tokens = tokenizer.tokenize(seq)
print(tokens)inputs = tokenizer(seq, return_tensors="pt")
inputs = {k: v.to(device) for k, v in inputs.items()}outputs = model(**inputs)
print(outputs.logits.shape)# 示例输出如下
"""
['M#', 'Ev', 'Vp', 'Qp', 'L#', 'Vy', 'Qd', 'Ya', 'Kv']
torch.Size([1, 11, 446])
"""(2)使用 esm 存储库加载 SaProt
用户也可以通过 esm 实现加载 SaProt。检查点存储在同一个 huggingface 文件夹中,名为 SaProt_650M_AF2.pt。作者提供了加载该模型的函数。
from utils.esm_loader import load_esm_saprotmodel_path = "/your/path/to/SaProt_650M_AF2.pt"
model, alphabet = load_esm_saprot(model_path)2、数据准备与处理
(1)将蛋白质结构转换为结构感知序列
研究提供了一个将蛋白质结构转换为结构感知序列的函数。该函数调用 foldseek 二进制文件对结构进行编码。
首先需要下载Foldseek工具:
-
从Google Drive链接下载foldseek二进制文件;
-
将文件放入项目的bin目录;
from utils.foldseek_util import get_struc_seq
pdb_path = "example/8ac8.cif"# Extract the "A" chain from the pdb file and encode it into a struc_seq
# pLDDT is used to mask low-confidence regions if "plddt_mask" is True. Please set it to True when
# use AF2 structures for best performance.
parsed_seqs = get_struc_seq("bin/foldseek", pdb_path, ["A"], plddt_mask=False)["A"]
seq, foldseek_seq, combined_seq = parsed_seqsprint(f"seq: {seq}")
print(f"foldseek_seq: {foldseek_seq}")
print(f"combined_seq: {combined_seq}")(2)获取训练数据
-
预训练数据集: 从HuggingFace下载
-
下游任务数据集: 从Google Drive下载
下载后将数据集解压到LMDB目录用于监督微调。
3、主要功能使用
(1)蛋白质表征提取
如果你想生成蛋白质嵌入,可以参考以下代码。嵌入是最后一层隐藏状态的平均值。
from model.saprot.base import SaprotBaseModel
from transformers import EsmTokenizerconfig = {"task": "base","config_path": "/your/path/to/SaProt_650M_AF2", # Note this is the directory path of SaProt, not the ".pt" file"load_pretrained": True,
}model = SaprotBaseModel(**config)
tokenizer = EsmTokenizer.from_pretrained(config["config_path"])device = "cuda"
model.to(device)seq = "M#EvVpQpL#VyQdYaKv" # Here "#" represents lower plDDT regions (plddt < 70)
tokens = tokenizer.tokenize(seq)
print(tokens)inputs = tokenizer(seq, return_tensors="pt")
inputs = {k: v.to(device) for k, v in inputs.items()}embeddings = model.get_hidden_states(inputs, reduction="mean")
print(embeddings[0].shape)(2)突变效应预测任务
研究提供了一个函数来预测蛋白质序列的突变效应。以下示例展示了如何预测特定位置的突变效应。如果使用 AF2 结构,作者强烈建议您添加 pLDDT 掩码(见下文)。
from model.saprot.saprot_foldseek_mutation_model import SaprotFoldseekMutationModelconfig = {"foldseek_path": None,"config_path": "/home/username/SaProt/model/Saprot/SaProt_650M_AF2", # Note this is the directory path of SaProt, not the ".pt" file"load_pretrained": True,
}
model = SaprotFoldseekMutationModel(**config)
tokenizer = model.tokenizerdevice = "cuda"
model.eval()
model.to(device)seq = "M#EvVpQpL#VyQdYaKv" # Here "#" represents lower plDDT regions (plddt < 70)# Predict the effect of mutating the 3rd amino acid to A
mut_info = "V3A"
mut_value = model.predict_mut(seq, mut_info)
print(mut_value)# Predict mutational effect of combinatorial mutations, e.g. mutating the 3rd amino acid to A and the 4th amino acid to M
mut_info = "V3A:Q4M"
mut_value = model.predict_mut(seq, mut_info)
print(mut_value)# Predict all effects of mutations at 3rd position
mut_pos = 3
mut_dict = model.predict_pos_mut(seq, mut_pos)
print(mut_dict)# Predict probabilities of all amino acids at 3rd position
mut_pos = 3
mut_dict = model.predict_pos_prob(seq, mut_pos)
print(mut_dict)(3)蛋白质逆折叠
from model.saprot.saprot_if_model import SaProtIFModel# Load model
config = {# Please download the weights from https://huggingface.co/westlake-repl/SaProt_650M_AF2_inverse_folding"config_path": "/your/path/to/SaProt_650M_AF2_inverse_folding","load_pretrained": True,
}device = "cuda"
model = SaProtIFModel(**config)
model = model.to(device)aa_seq = "##########" # All masked amino acids will be predicted. You could also partially mask the amino acids.
struc_seq = "dddddddddd"# Predict amino acids given the structure sequence
pred_aa_seq = model.predict(aa_seq, struc_seq)
print(pred_aa_seq)4、模型训练与评估
(1)模型微调
研究提供了一个在数据集上微调 SaProt 的脚本。下面的代码展示了如何对特定下游任务的 SaProt 进行微调。运行代码前,请确保数据集放在 LMDB 文件夹中,SaProt 650M 模型的 huggingface 版本放在 weights/PLMs 文件夹中。请注意,由于不同用户的硬件限制,默认训练设置与论文中的不尽相同。我们建议用户根据自身条件(如 batch_size、devices 和 accumulate_grad_batches)灵活修改 yaml 文件。
提供了针对不同下游任务的配置文件:
# 在热稳定性任务上微调SaProt
python scripts/training.py -c config/Thermostability/saprot.yaml# 在热稳定性任务上微调ESM-2
python scripts/training.py -c config/Thermostability/esm2.yaml(2)零样本性能评估
研究提供了一个脚本来评估模型的零样本性能(需要将foldseek二进制文件放在bin文件夹中):
# 1、评估SaProt在ProteinGym基准上的零样本性能,结果将保存在 output/ProteinGym 文件夹中。
python scripts/mutation_zeroshot.py -c config/ProteinGym/saprot.yaml# 2、评估ESM2在ProteinGym基准上的零样本性能
python scripts/mutation_zeroshot.py -c config/ProteinGym/esm2.yaml# 3、评估ClinVar基准性能,可以使用以下脚本来计算 AUC 指标
python scripts/mutation_zeroshot.py -c config/ClinVar/saprot.yaml
python scripts/compute_clinvar_auc.py -c config/ClinVar/saprot.yaml5、注意事项
(1)pLDDT遮蔽: 使用AF2结构时建议开启pLDDT遮蔽(plddt_mask=True)以获得最佳性能
(2)训练配置: 默认训练配置可能需要根据硬件条件调整,主要包括:
batch_size
devices
accumulate_grad_batches
(3)Wandb记录: 如需记录训练过程:
修改配置文件 Trainer.logger = True
在setting.os_environ.WANDB_API_KEY中添加wandb API密钥
参考文档:
https://github.com/westlake-repl/SaProt
SaProt: Protein Language Modeling with Structure-aware Vocabulary | OpenReview