sc-atac的基础知识(0)
sc-atac的基础知识
**fragment**是ATAC-seq实验中的一个重要概念,它指的是通过Tn5转座酶对DNA分子进行酶切,然后经由双端测序得到的序列。
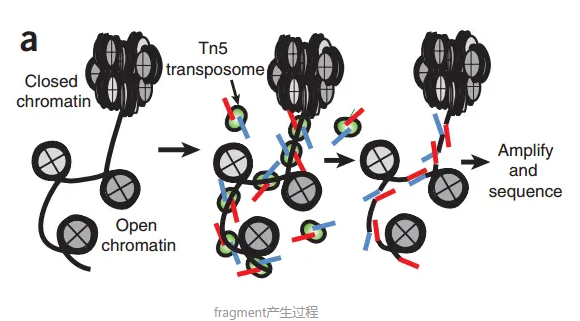
根据Tn5插入导致的偏移从read比对得到的位置推断出fragment的起始和结束位置。根据之前的报道,Tn5转座酶以同源二聚体的形式结合到DNA上,在两个Tn5分子间隔着9-bp的DNA序列。根据这个情况,每个Tn5同源二聚体的结合事件会产生2个Insertions,中间隔着9bp。因此,真实的"开放"位置的中心在Tn5二聚体的正中间,而不是Tn5的插入位置。为了尽可能的还原真实情况,我们对Tn5的Insertions进行了校正,即正链的插入结果往右移动4bp(+4 bp), 负链的插入结果往左偏移5bp(-5 bp)。这个和最早提出的ATAC-seq里的描述是一致的。因此,在ArchR中,"fragment"指的是一个table或genomic ranges对象, 记录在染色体上,经过偏移校正后的单碱基起始位置,以及经过偏移校正后单碱基结束位置,每个fragment都会对应唯一的细胞条形码。类似的,"Insertions"这得是偏移校正后的单碱基位置,它位于开放位置的正中心。(https://www.jianshu.com/p/f7975da8e1e8)
- Tn5 转座酶(绿色剪刀图标)只能切割 开放染色质(Open chromatin,线条松散)。
- 在封闭染色质(Closed chromatin,线条紧密)处,Tn5 无法进入,因此不切割。
- Tn5 在开放区域切割后,插入测序接头(adapters,灰色小条),形成 可测序的 DNA 片段。
- 这些片段经过 PCR 扩增 & 测序,最终得到 开放染色质信号峰。
“只有染色质松开的区域才会被 Tn5 切割并变成可测序的 ATAC 片段。”
Tn5 转座酶是同源二聚体
什么是同源二聚体,组蛋白是八聚体吗?两者什么区别
一句话区分:
- 同源二聚体 = 2 个相同蛋白抱在一起;
- 组蛋白八聚体 = 8 个不同组蛋白(2×H2A、2×H2B、2×H3、2×H4)按固定比例拼成核小体核心。
1 同源二聚体(homo-dimer)
- 定义:由 两条完全相同的肽链/蛋白 非共价结合而成的功能单位。
- ATAC-seq 场景:Tn5 转座酶只有 两条一模一样的 Tn5 分子 同时结合 DNA,才能切割并插入接头。
2 组蛋白八聚体(histone octamer)
- 组成:
(H2A-H2B) × 2 (H3-H4) × 2 共 8 条肽链 - 功能:绕成核小体核心,把 DNA 缠成“线轴”。
3 核心差异
| 维度 | 同源二聚体 | 组蛋白八聚体 |
|---|---|---|
| 链数 | 2 条相同蛋白 | 8 条不同蛋白 |
| 蛋白种类 | 1 种 | 4 种(H2A、H2B、H3、H4) |
| 生物学角色 | 酶活性单位(如 Tn5) | 染色质结构骨架 |
| 是否完全相同 | ✅ 是 | ❌ 否 |
根据之前的报道,Tn5转座酶以同源二聚体的形式结合到DNA上,在两个Tn5分子间隔着9-bp的DNA序列。什么意思
意思是:
Tn5 转座酶只有两条完全相同的酶链(同源二聚体)同时抓住 DNA,它们之间被 9 个碱基对(9 bp)的 DNA 片段隔开;
- 正链酶切在 +4 bp 处
- 负链酶切在 -5 bp 处
校正后,这两个切点中间正好留下 9 bp 的“间隙”,真正的开放染色质中心就在这段间隙的正中间。
根据这个情况,每个Tn5同源二聚体的结合事件会产生2个Insertions,中间隔着9bp。什么是Insertions
在 ATAC-seq 里,Insertion 指的是 Tn5 转座酶在 DNA 上实际切割并把测序接头“插入”进去的那一个单碱基点。
- 一个 Tn5 同源二聚体结合事件会 同时切两下,因此 产生 2 个这样的插入点(两条链各一个)。
- 这两个插入点之间正好隔着 9 bp 的 DNA 片段。
Insertions与Fragment的区别
-
Insertions 是 单个碱基点(Tn5 二聚体中心),
例如 chr1:1000(正链 +4 校正后)。 -
Fragment 是 一条线段(起点—终点),
例如 chr1:1000–1001(起点 1000,终点 1001),长度 1 bp,正好以 Insertion 点为中心。
因此,Insertion 位于 Fragment 的正中间,两者校正后的坐标只差一个“线段宽度”,方向相反的两端各 1 bp。
为什么使用ArchR
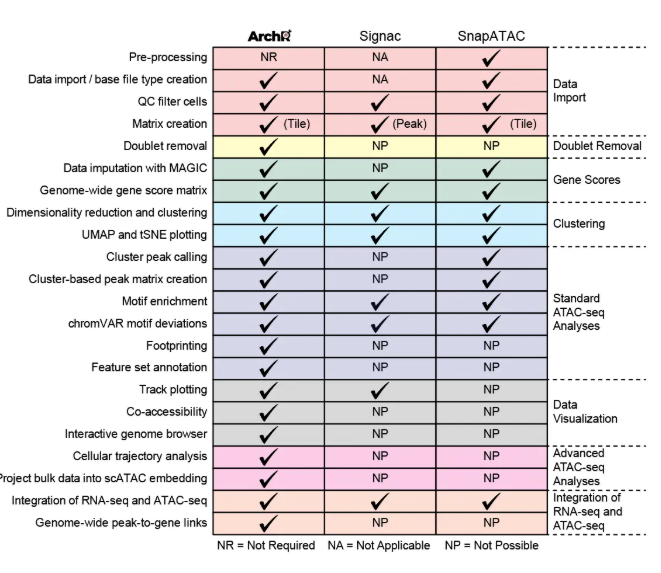
- 行标题:表示不同的功能模块,如数据导入(Data import)、双细胞去除(Doublet Removal)、基因得分(Gene Scores)、聚类(Clustering)、标准ATAC-seq分析(Standard ATAC-seq Analyses)、数据可视化(Data Visualization)和高级ATAC-seq分析(Advanced ATAC-seq Analyses)等。
ArchR通过优化数据结构降低了内存消耗,使用并行提高了运行速度,因此保证其性能优于其他同类型工具
什么是Arrow文件/ArchRProject
proj <- ArchRProject(ArrowFiles = ArrowFiles,genomeAnnotation = genomeAnnotation,geneAnnotation = geneAnnotation,copyArrows = TRUE
)
- Arrow 文件的作用
- Arrow 文件:是 ArchR 项目中用于存储每个独立样本所有相关信息的文件格式。这些信息包括元数据(如样本名称、实验条件等)、开放的染色质片段(fragments)以及数据矩阵等。
- 独立样本:每个 Arrow 文件对应一个独立样本,这个样本最好是详尽的分析单元,例如在特定实验条件下的单个重复实验。
- Arrow 文件的内容
- 文件内容:Arrow 文件记录了每个样本的所有相关信息,这些信息对于后续的分析至关重要。
- 文件路径:Arrow 文件实际上是存储在磁盘上的 HDF5 文件,而不是内存中的 R 对象。
·Arrow 文件:存储在磁盘上的文件,使用 HDF5 格式,可以存储大量数据,并且支持快速读取和写入。
·R 对象:存储在内存中的数据结构,用于数据处理和分析,但不适合存储大量数据,因为内存容量有限。
- ArchR 处理 Arrow 文件的方式
- 编辑和更新:在创建 Arrow 文件以及进行一些附加分析时,ArchR 会编辑和更新相应的 Arrow 文件,添加额外的信息层。
- 高效访问:通过使用 ArchRProject 对象,ArchR 可以将多个 Arrow 文件关联到一个分析框架中,从而确保在 R 中能高效访问这些文件。
- ArchRProject 对象的作用
- 内存占用:ArchRProject 对象本身占用的内存不多,因为它主要负责关联磁盘上的 Arrow 文件,而不是存储文件内容本身。
- 分析框架:ArchRProject 对象将多个 Arrow 文件关联到一个分析框架中,使得在 R 中可以高效地访问和处理这些文件。
- 总结
- Arrow 文件:存储每个样本所有相关信息的 HDF5 文件,存储在磁盘上。
- ArchRProject 对象:用于关联多个 Arrow 文件,使其在 R 中高效访问,占用内存少。
- 处理流程:在创建和分析过程中,ArchR 会更新 Arrow 文件,添加额外信息层,以支持复杂的数据分析。
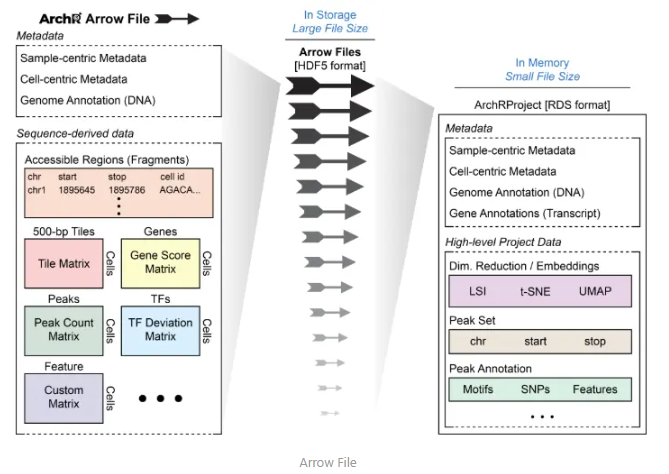
这张图展示了 ArchR 项目中== Arrow 文件==和 ArchRProject 对象的结构和内容,以及它们在存储和内存中的不同格式。
左侧:Arrow 文件(存储在磁盘上)
- 格式:HDF5 格式,文件体积较大。
- 内容:
- Metadata(元数据):
- 样本中心元数据(Sample-centric Metadata)
- 细胞中心元数据(Cell-centric Metadata)
- 基因组注释(Genome Annotation,DNA)
- Sequence-derived data(序列衍生数据):
- 可访问区域(Accessible Regions,Fragments)
- 500 bp 的片段(Tiles)
- 基因得分矩阵(Gene Score Matrix)
- 峰值(Peaks)
- 峰值计数矩阵(Peak Count Matrix)
- 转录因子偏差矩阵(TF Deviation Matrix)
- 细胞类型矩阵(Cell Type Matrix)
- Metadata(元数据):
右侧:ArchRProject 对象(加载到内存中)
- 格式:RDS 格式,文件体积较小。
- 内容:
- Metadata(元数据):
- 样本中心元数据(Sample-centric Metadata)
- 细胞中心元数据(Cell-centric Metadata)
- 基因组注释(Genome Annotation,DNA 和转录本)
- High-level Project Data(高级项目数据):
- 降维/嵌入(Dim. Reduction / Embeddings,如 LSI、t-SNE、UMAP)
- 峰值集合(Peak Set,包括染色体位置、起始、终止位置)
- 峰值注释(Peak Annotation,包括 Motifs、SNPs、Features)
- Metadata(元数据):
总结
- Arrow 文件存储了单细胞 ATAC-seq 数据的原始信息,包括元数据和序列衍生数据,文件体积较大,存储在磁盘上。
- ArchRProject 对象是一个内存中的对象,包含了 Arrow 文件的索引和一些高级分析数据,文件体积较小,便于在内存中快速访问和处理。
- 通过使用 ArchRProject 对象,可以高效地访问和管理磁盘上的 Arrow 文件,同时利用内存中的对象进行快速分析。
有一些操作会直接修改Arrow文件,而一些操作会先作用于ArchRproject,接着反过来更新每个相关Arrow文件。因为Arrow文件是以非常大的HDF5格式存放,所以ArchR的get-er函数通过和ArchRProject进行交互获取数据,而add-er函数既能直接在Arrow文件中添加数据,也能直接在ArchRpoject里添加数据,或者通过ArchRpoject向Arrow文件添加数据。