【从GEO数据库批量下载数据】
从GEO数据库批量下载数据
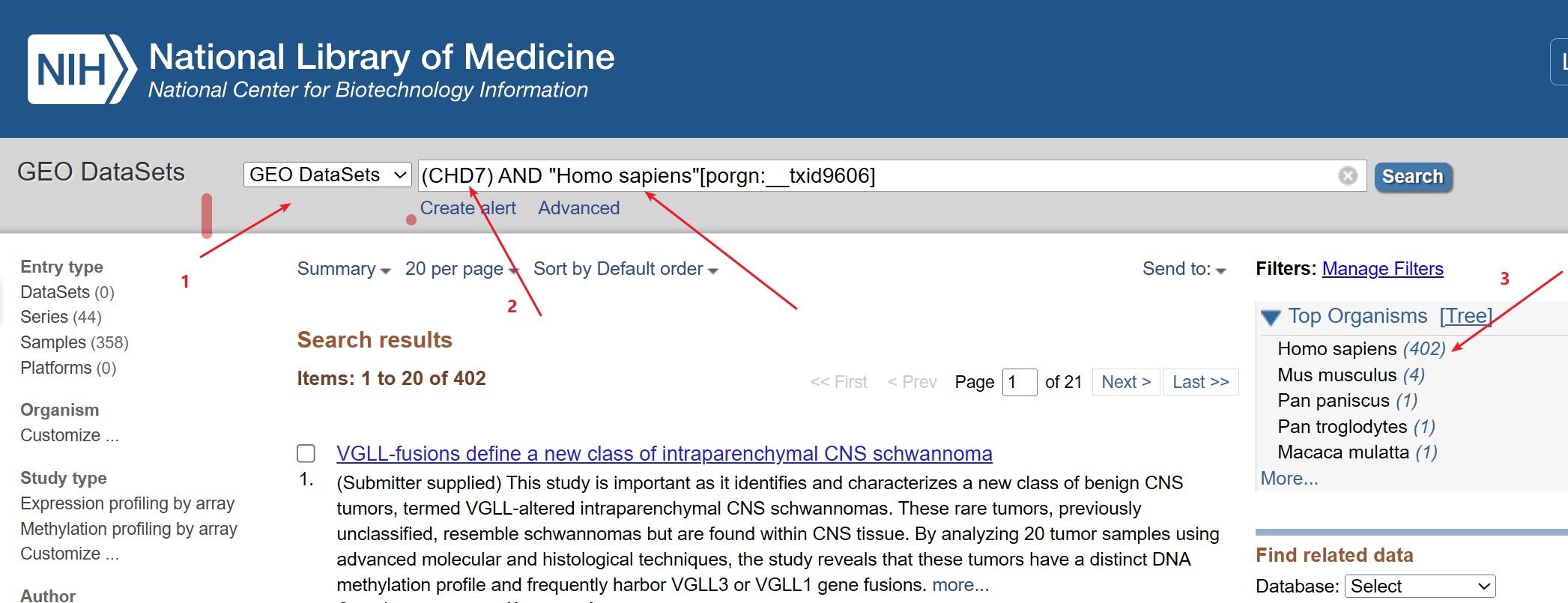
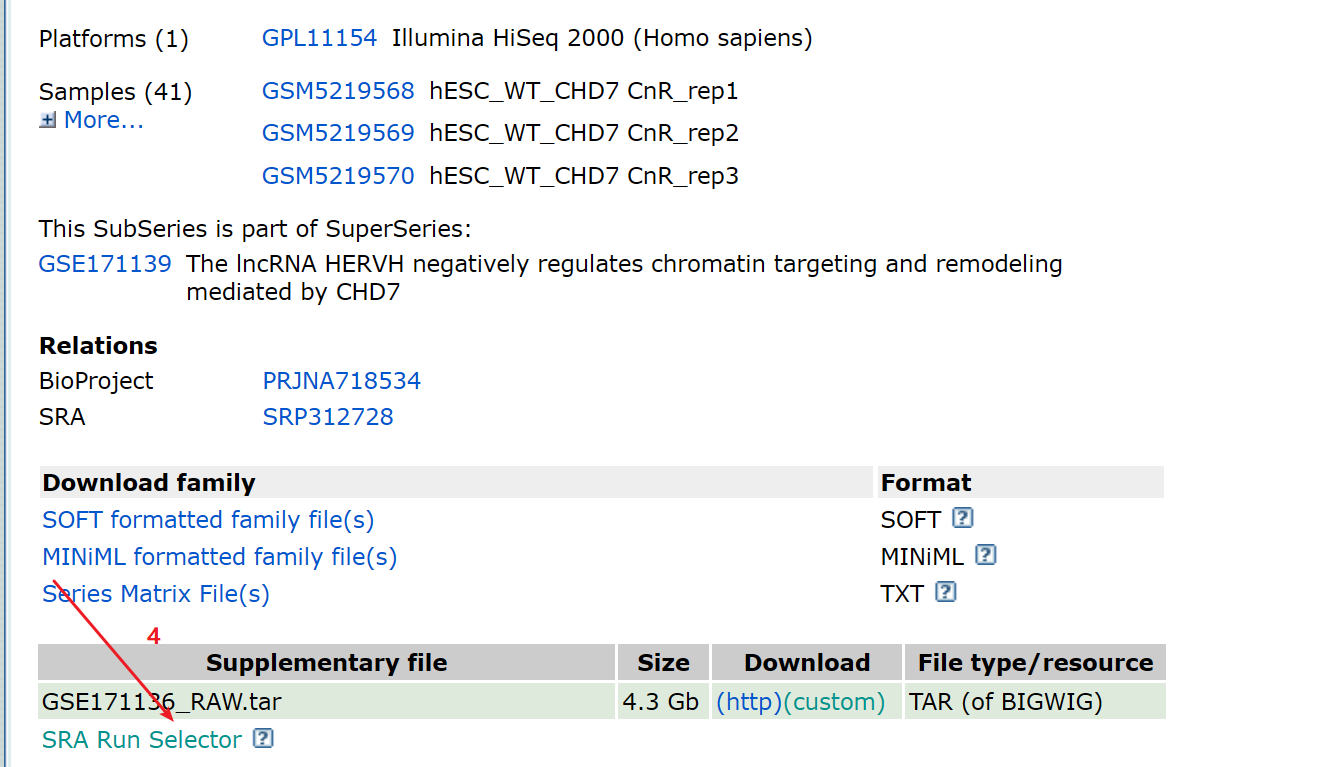
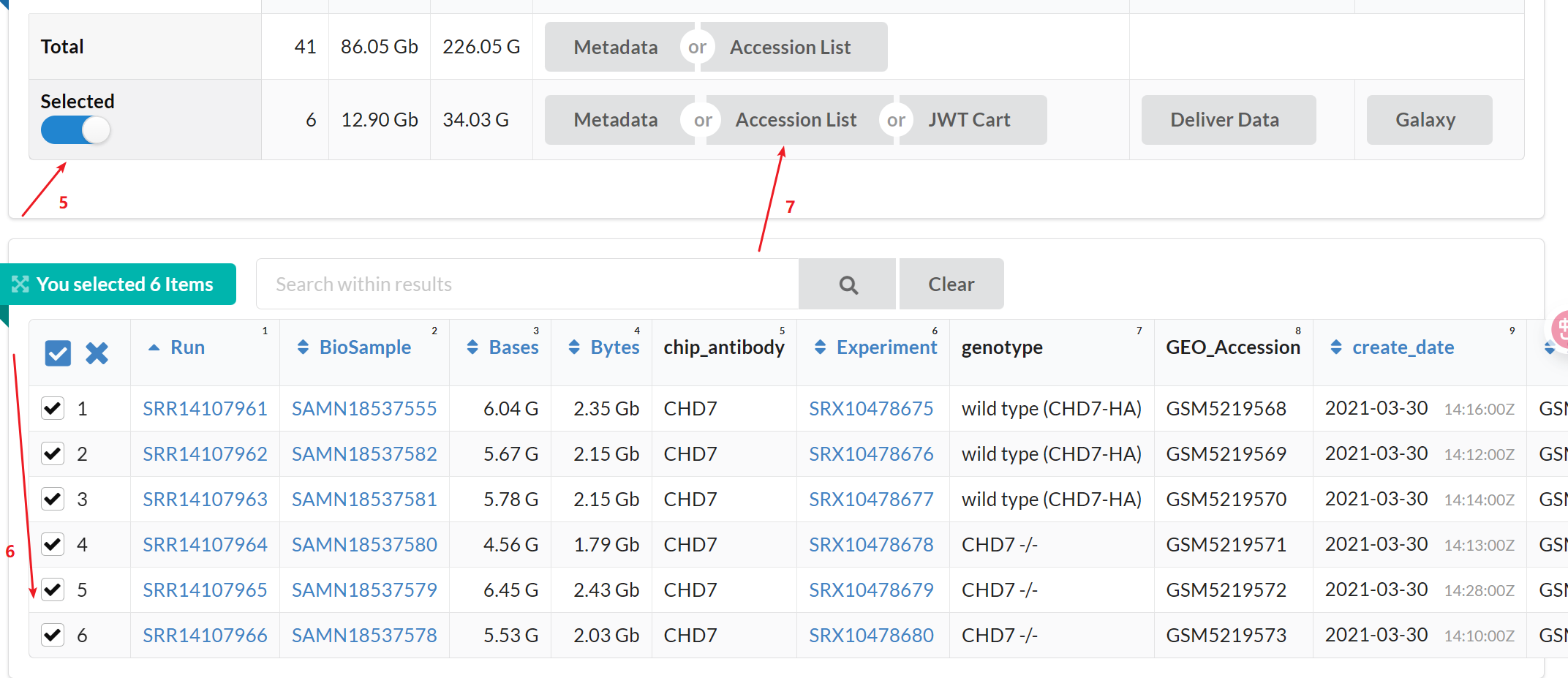
1:进入GEO DataSets拿到所需要下载的数据的srr.list,上传到linux, 就可以使用prefetch这个函数来下载
2:操作步骤如下:
conda 安装sra-tools
conda create -n sra-env -c bioconda -c conda-forge sra-tools=3.0.7 -y
conda activate sra-env
which prefetch
>> /data/user/yzhao/miniconda3/envs/sra-env/bin/prefetch
下载
#组织好自己的数据,先进入文件夹
cd /data/user/yzhao/project/2025_hCHD7
mkdir {sra,raw,clean,align,peaks,motif,qc}
cd sra
#prefetch下载数据,SRR_Acc_List.txt就是我命名的包含 SRR ID 列表的文件。也就是srr.list
cat ../rawdata/SRR_Acc_List.txt | while read id; do echo "Downloading $id..."; nohup prefetch "$id" --max-size 100G -O ./ & done
#实时查看 nohup.out 文件的最新输出内容,通常用于观察后台运行程序的日志,按 Ctrl + C 即可退出 tail -f 实时查看模式。
tail -f nohup.out
#漏下了一个,可用prefetch单独下载
prefetch SRR14107966 -O /data/user/yzhao/project/2025_hCHD7/sra/sra/
#方法1:得到fastq测序数据
vim sra2fastq.sh
#!/bin/bashTHREADS=8
INDIR="/data/user/yzhao/project/2025_hCHD7/sra/sra"
OUTDIR="/data/user/yzhao/project/2025_hCHD7/raw"mkdir -p "$OUTDIR"for sra_dir in "$INDIR"/*; doif [ -d "$sra_dir" ]; thensample=$(basename "$sra_dir")sra_file="$sra_dir/$sample.sra"if [ -f "$sra_file" ]; thenecho "Converting $sample ..."fasterq-dump "$sra_file" -e $THREADS -O "$OUTDIR"# 压缩fastq文件if [ -f "$OUTDIR/${sample}_1.fastq" ]; thengzip "$OUTDIR/${sample}_1.fastq"fiif [ -f "$OUTDIR/${sample}_2.fastq" ]; thengzip "$OUTDIR/${sample}_2.fastq"fiif [ -f "$OUTDIR/${sample}.fastq" ]; thengzip "$OUTDIR/${sample}.fastq"fielseecho "Warning: $sra_file not found!"fifi
done
chmod +x sra2fastq.sh
nohup bash sra2fastq.sh >sra2fastq.sh.log &# 方法2:在获得fastq的过程中完成样品名称的替换
vim sra2fastq.sh2
#!/bin/bashTHREADS=8
INDIR="/data/user/yzhao/project/2025_hCHD7/sra/sra"
OUTDIR="/data/user/yzhao/project/2025_hCHD7/raw"# 建立 SRR -> 样本名 的映射
declare -A NAME_MAP=([SRR14107961]=CHD7_HA1[SRR14107962]=CHD7_HA2[SRR14107963]=CHD7_HA3[SRR14107964]=KO1[SRR14107965]=KO2[SRR14107966]=KO3
)for sra_dir in "$INDIR"/*; doif [ -d "$sra_dir" ]; thensrr_id=$(basename "$sra_dir")sra_file="$sra_dir/$srr_id.sra"# 判断文件是否存在并在映射表中if [ -f "$sra_file" ] && [[ ${NAME_MAP[$srr_id]+_} ]]; thennew_name=${NAME_MAP[$srr_id]}echo "Converting $srr_id to $new_name ..."# 转换为 fastq(会生成 _1.fastq 和 _2.fastq)fasterq-dump "$sra_file" -e $THREADS -O "$OUTDIR"# 重命名并压缩if [ -f "$OUTDIR/${srr_id}_1.fastq" ]; thenmv "$OUTDIR/${srr_id}_1.fastq" "$OUTDIR/${new_name}_1.fastq"gzip "$OUTDIR/${new_name}_1.fastq"fiif [ -f "$OUTDIR/${srr_id}_2.fastq" ]; thenmv "$OUTDIR/${srr_id}_2.fastq" "$OUTDIR/${new_name}_2.fastq"gzip "$OUTDIR/${new_name}_2.fastq"fiif [ -f "$OUTDIR/${srr_id}.fastq" ]; thenmv "$OUTDIR/${srr_id}.fastq" "$OUTDIR/${new_name}.fastq"gzip "$OUTDIR/${new_name}.fastq"fielseecho "Warning: $sra_file not found or no name mapping!"fifi
done
chmod +x sra2fastq.sh2
nohup bash sra2fastq.sh2 > sra2fastq.sh2.log 2>&1 &