单细胞测序细胞注释全攻略:选择自动工还是手动验证,附常见细胞Marker基因
近年来,单细胞测序技术在生物医学研究中迅速发展,揭示了生命活动前所未有的细节。然而,面对浩如烟海的单细胞数据,如何准确地给细胞群体贴上“身份证”——即细胞类型注释,成为了研究者的关键一步。
本文将详细讲解单细胞测序中细胞类型注释的两大策略:自动工具与手动验证,并附上常用细胞Marker基因,助你轻松入门单细胞测序数据分析。
一、自动工具注释:高效又省心
自动注释工具可以大大提升分析效率,常用的工具包括:
| 工具名称 | 注释原理 | 特点 |
|---|---|---|
| SingleR | 利用参考转录组数据,通过相关性自动注释 | 便捷快速,依赖参考数据 |
| scCATCH | 依赖CellMarker数据库,快速自动注释 | 数据库依赖性强 |
| CellTypist | 基于机器学习模型和预训练数据进行细胞分类 | 精准高效,需预训练模型 |
优点:节省时间,快速得到初步结果。
缺点:依赖于参考数据库的完整性,可能忽略稀有或新发现的细胞类型。
实操演示(以Seurat为例)
# 标准注释流程代码示例pbmc <- CreateSeuratObject(counts = pbmc_data)pbmc <- NormalizeData(pbmc)pbmc <- FindVariableFeatures(pbmc)pbmc <- ScaleData(pbmc)pbmc <- RunPCA(pbmc)pbmc <- FindNeighbors(pbmc, dims = 1:10)pbmc <- FindClusters(pbmc, resolution = 0.5)pbmc <- RunUMAP(pbmc, dims = 1:10)# 使用SingleR自动注释library(SingleR)pred <- SingleR(test = pbmc@assays$RNA@data,ref = HumanPrimaryCellAtlasData(),labels = HumanPrimaryCellAtlasData()$label.main)
二、手动验证注释:精准又可靠
手动验证一般基于经典的Marker基因进行验证。研究者需要具备一定的生物学背景,参考文献与数据库对细胞类型进行核对。
手动验证一般基于经典的Marker基因进行验证。这一过程包括以下步骤:
- 初步筛选Marker基因:研究者根据既往文献、公共数据库(如CellMarker、PanglaoDB)确定目标细胞类型的经典标志基因。
- 数据可视化与验证:使用Seurat、Scanpy等工具的UMAP、t-SNE或DotPlot等方法进行Marker基因的表达可视化,观察目标基因在细胞群中的特异性表达情况。
- 多基因交叉验证:为提高准确性,通常需要选取多个Marker基因进行交叉验证,从而更准确地定义细胞亚型。
- 文献与数据库核实:进一步查阅最新文献和权威数据库,确认所注释细胞类型与已报道的细胞特征是否一致。
常用的细胞类型Marker基因列表如下:
1. 免疫系统细胞
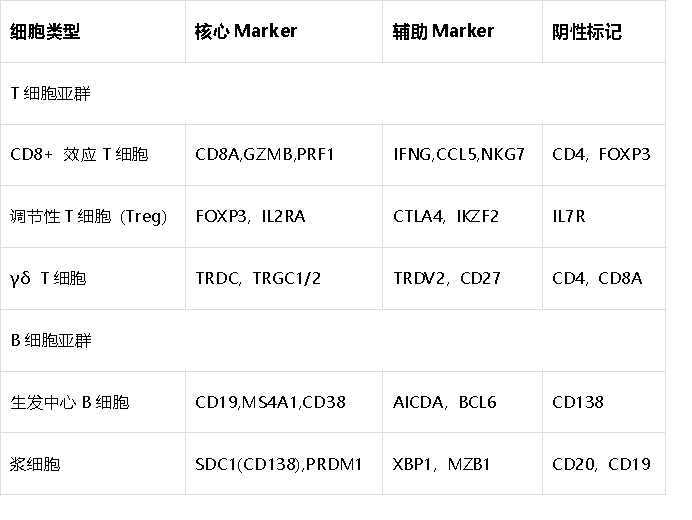
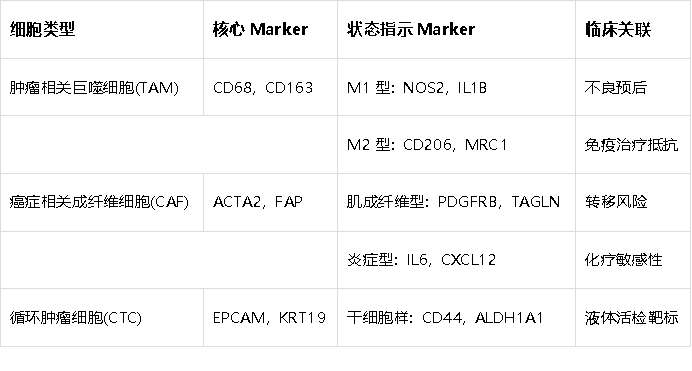
2. 肿瘤微环境细胞
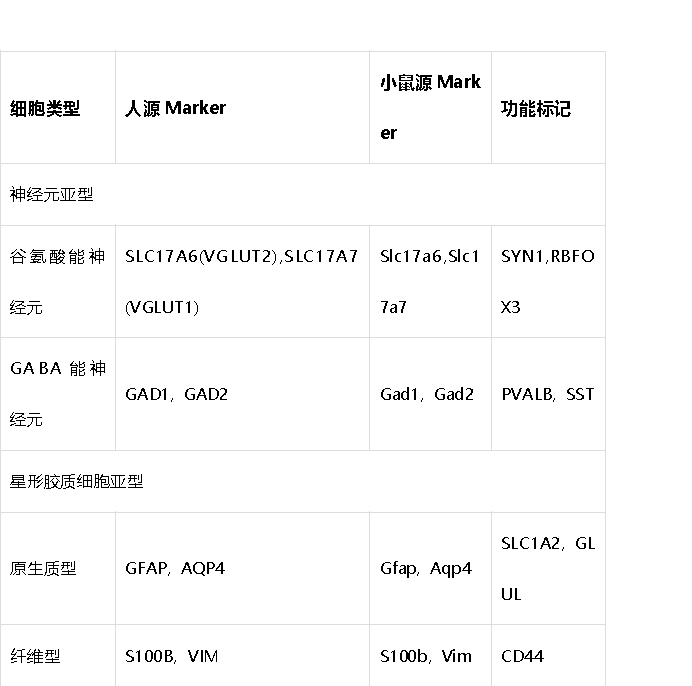
3. 神经系统细胞
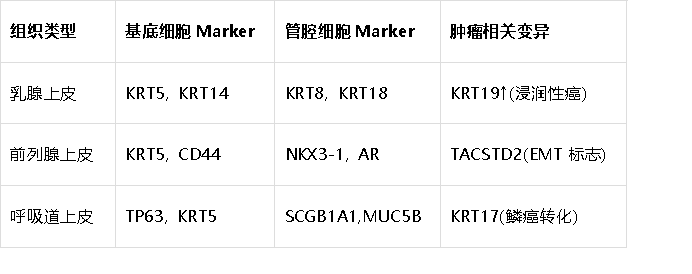
4. 上皮组织细胞
5. 稀有细胞类型
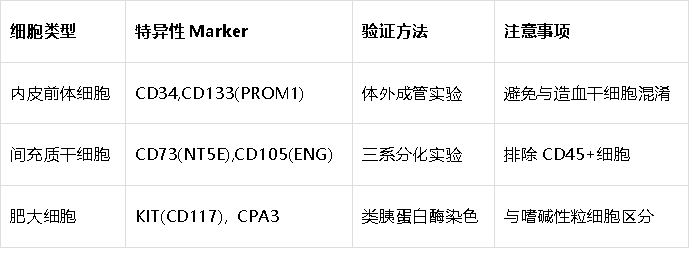
数据获取方式
1. 动态数据库:
CellMarker 3.0
Allen Brain Atlas(神经特异性)
2. 本地资源包
# 下载预编译Marker基因集wget https://singlecellmarkers.org/human_all.csv
三、组合策略:推荐的实践方法
实际研究中,推荐的细胞注释策略是自动注释与手动验证相结合。
推荐流程:
- 利用自动工具(如SingleR、CellTypist)进行初步快速注释。
- 根据自动注释结果和经典Marker基因,进行手动验证与修正。
- 必要时,借助最新的大语言模型(如ChatGPT、GeneGPT等)进一步交叉验证和功能解读。
通过上述组合策略,研究人员既能高效处理数据,又能确保结果的准确性和可靠性。
小结
细胞类型注释的质量决定了单细胞测序后续分析的深度与准确性。自动工具与手动验证各有优势,灵活运用二者的组合策略才能游刃有余。希望本文的攻略与细胞Marker基因列表,能帮助大家在单细胞测序领域迈出坚实一步!
关注我们,获取更多前沿生物信息学研究成果!有什么想法可以在评论区评论,也可以私信获取原文PDF哦!
