SnakeMake搭建pipeline 1
https://snakemake.readthedocs.io/en/stable/tutorial/tutorial.html
首先是安装:
mamba create -c conda-forge -c bioconda -y -n snakemake snakemake
下面是snakemake中的示例教程:

然后按照对应的教程一步一步进行示意操作
当然还可以集成到本地主机vscode中进行演示:



1,简单rule

这是我们在工作目录中的文件组织:


上面是我们按照指南指示创建的新文件


输出效果如下:只是演示的效果,并不是实际执行的过程


当然我们可以实际执行这个命令:



2,泛化之后的rule:
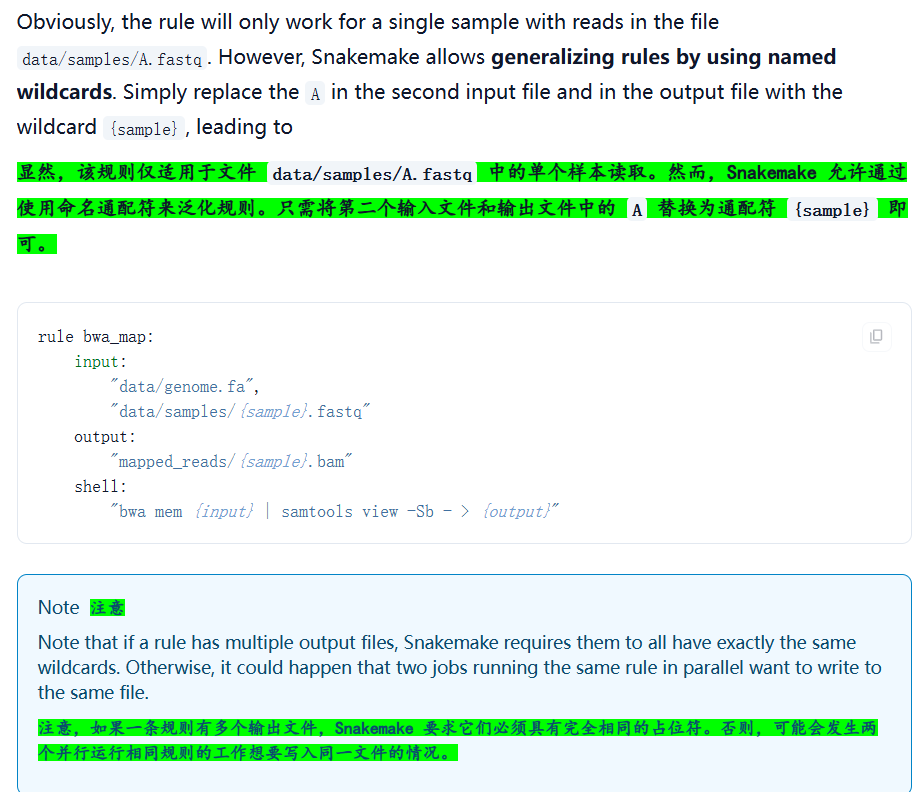
现在修改我们的rule文件:









3,开始增加规则:

在 Snakemake 中,wildcards 是一个非常重要的概念,它允许规则动态地处理多个输入文件,而不需要硬编码文件名。下面我将详细解释 wildcards 的含义,并通过一个简单的例子来说明它的使用。
1. 什么是 wildcards?
在 Snakemake 中,wildcards 是一种动态匹配机制,用于在规则中处理多个类似的文件。它允许你在规则的输入、输出和命令中使用通配符(wildcards),这些通配符会在运行时被具体的值替换。
- 通配符(wildcards):通常用
{}表示,例如{sample}。它是一个占位符,用于匹配文件名中的某个部分。 wildcards** 对象**:在规则的执行过程中,Snakemake 会根据输入和输出文件的模式,自动解析出通配符的具体值,并将这些值存储在wildcards对象中。你可以在规则的命令部分通过wildcards.sample等方式访问这些值。
2. 简单例子:对多个样本的文件进行排序
假设你有多个样本的比对结果文件,文件名格式为 mapped_reads/{sample}.bam,你希望对每个样本的文件进行排序,并将排序后的文件存储到 sorted_reads/{sample}.sorted.bam。
Snakemake 规则示例
rule sort_bam:
input:
"mapped_reads/{sample}.bam" # 输入文件模式
output:
"sorted_reads/{sample}.sorted.bam" # 输出文件模式
shell:
"samtools sort -T {wildcards.sample} -o {output} {input}"
- 输入文件模式:
mapped_reads/{sample}.bam:这里{sample}是一个通配符,表示样本的名称。Snakemake 会自动匹配所有符合这个模式的文件。例如,如果目录中有mapped_reads/sample1.bam和mapped_reads/sample2.bam,那么{sample}会被解析为sample1和sample2。
- 输出文件模式:
sorted_reads/{sample}.sorted.bam:输出文件的名称也包含通配符{sample}。Snakemake 会根据输入文件中解析出的{sample}值,生成对应的输出文件名。例如,对于输入文件mapped_reads/sample1.bam,输出文件将是sorted_reads/sample1.sorted.bam。
wildcards** 对象**:- 在
shell命令中,{wildcards.sample}被用来访问通配符{sample}的值。这个值是从输入文件名中解析出来的。 - 例如:
- 如果输入文件是
mapped_reads/sample1.bam,那么{wildcards.sample}的值就是sample1。 - 如果输入文件是
mapped_reads/sample2.bam,那么{wildcards.sample}的值就是sample2。
- 如果输入文件是
- 在
samtools sort** 命令**:samtools sort -T {wildcards.sample} -o {output} {input}:-T {wildcards.sample}:指定临时文件的前缀为样本名称(sample1或sample2)。-o {output}:指定输出文件路径。{input}:指定输入文件路径。
实际运行过程
假设目录结构如下:
mapped_reads/
├── sample1.bam
├── sample2.bam
Snakemake 会自动解析出两个任务:
- 对
mapped_reads/sample1.bam进行排序,生成sorted_reads/sample1.sorted.bam。- 命令:
samtools sort -T sample1 -o sorted_reads/sample1.sorted.bam mapped_reads/sample1.bam
- 命令:
- 对
mapped_reads/sample2.bam进行排序,生成sorted_reads/sample2.sorted.bam。- 命令:
samtools sort -T sample2 -o sorted_reads/sample2.sorted.bam mapped_reads/sample2.bam
- 命令:
3. 总结
wildcards** 的作用**:它允许你在规则中使用动态的文件名模式,而不是硬编码具体的文件名。这样可以让你的规则更通用,能够处理多个类似的文件。wildcards** 对象**:在规则的命令部分,可以通过{wildcards.<wildcard_name>}的方式访问通配符的具体值,从而动态地生成命令。
总之我们增加了新规则:



再次增加更新新规则:











4,在规则中使用自定义的脚本,而不是调用现有的工具






也可以实际执行一下:



5,添加目标规则:





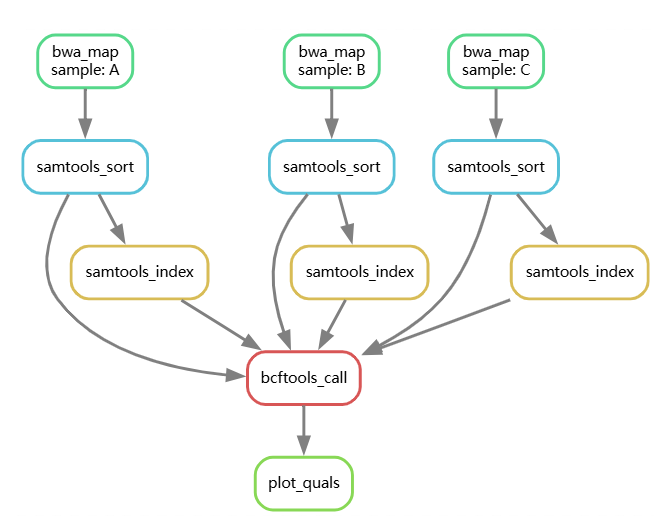
总之,目前的流程如下:
SAMPLES = ["A", "B"]
rule all:
input:
"plots/quals.svg"
rule bwa_map:
input:
"data/genome.fa",
"data/samples/{sample}.fastq"
output:
"mapped_reads/{sample}.bam"
shell:
"bwa mem {input} | samtools view -Sb - > {output}"
rule samtools_sort:
input:
"mapped_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam"
shell:
"samtools sort -T sorted_reads/{wildcards.sample} "
"-O bam {input} > {output}"
rule samtools_index:
input:
"sorted_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam.bai"
shell:
"samtools index {input}"
rule bcftools_call:
input:
fa="data/genome.fa",
bam=expand("sorted_reads/{sample}.bam", sample=SAMPLES),
bai=expand("sorted_reads/{sample}.bam.bai", sample=SAMPLES)
output:
"calls/all.vcf"
shell:
"bcftools mpileup -f {input.fa} {input.bam} | "
"bcftools call -mv - > {output}"
rule plot_quals:
input:
"calls/all.vcf"
output:
"plots/quals.svg"
script:
"scripts/plot-quals.py"