分子动力学软件包Amber24的安装
AmberTools24 概述
AmberTools 是一个用于分子模拟的软件套件,提供了一系列用于模拟辅助、建模分析以及可视化工具。AmberTools 最初是为了支持 Amber 分子模拟软件包的开发而发布的,在分子动力学模拟、药物设计和生物信息学等领域中被广泛使用。
AmberTools24(2024年4月30日发布)由以下主要代码组成:
- antechamber和MCPB.py:为一般有机分子和金属中心创建力场的程序
- tleap和parmed:Amber 模拟的基本准备工具
- sqm和Quick:半经验、DFTB 和从头算量子化学代码,用于独立计算或 QM/MM
- pbsa:对泊松-玻尔兹曼模型进行数值解
- 3D-RISM:解决积分方程模型以进行溶解
- sander:分子动力学模拟的主力程序
- gem.pmemd:使用高级力场的工具
- mdgx:一款主要通过参数拟合来突破 Amber MD 界限的程序。还包括可定制的虚拟站点和显式溶剂 MD 功能。
- cpptraj和pytraj:用于分析轨迹结构和动态的工具
- mmpbsa.py:基于能量的 MD 轨迹分析
- FEW:自由能量工作台,提供各种自由能量分析工具
- fe-toolkit:分析炼金术自由能模拟的例程
AmberTools24新功能:
- 新的和更新的力场:纳入 GAFF2 的 ABCG2 电荷模型
- 用于 Hartree-Fock 和 DFT 电子结构计算的快速软件包,具有 GPU 支持。Quick 集成到 _sander_中用于 QM/MM 模拟,AmberTools24 包含显著的性能改进、对 f 基函数的支持、更新的几何优化器以及对自旋无限制计算的支持。
- cpptraj 更新: (另请参阅cpptraj 的详细更新日志。)
- 修复了 NPT 轨迹的展开和扩散成像。
- 增加了展开 NPT 轨迹时消除盒子波动的能力(通过投影到平均单元格上)。
- 添加了来自多个时间原点的 MPI 并行扩散计算,以及环面视图保留扩散计算。
- 对于核酸结构分析,增加了写入轴、采用用户指定的碱基对以及报告所有氢键(不仅仅是 WCF)的能力。
- 增加了使用 OpenMM 从能量命令计算总能量的功能(如果链接到 OpenMM 库)。
- 增加了改变选定原子的质量和/或电荷的能力。
- 增加了计算轨迹中结构扩展相似度得分的功能(J. Comp.-Aid. Mol. Design, 2022, 36, 157-173)。
- 增加了利用和操纵内部坐标(Z 矩阵)的能力。
- 向 GIST 添加了 MPI 并行性和可旋转网格。
- 在‘change’命令中添加了‘mergeres’关键字,用于合并连续残基。
- 增加了在“atommap”命令中将映射原子名称更改为引用名称的功能。
- 在‘box’命令中添加了‘getbox’关键字,以允许提取单元格、分数单元格或对称形状矩阵数据。
- 添加了“--charge”命令行标志,用于计算电荷并打印到标准输出。
- fe-toolkit更新了用于分析炼金术自由能计算的例程包。
Amber24 概述
Amber24 软件包在 AmberTools24 基础上添加了pmemd 程序,该程序类似于AmberTools 中的sander(分子动力学)代码,但在多个 CPU 上提供了(更)更好的性能,并在 GPU 上显著提高了速度。
Amber24主要新功能包括:
- 对 AMD GPU 的一般支持。
- 炼金术transformation pathways/softcore potentials和炼金术增强采样 (ACES) 方法的提升:
- 高级 lambda 调度选项,包括用于优化相空间重叠 lambda 间距的新工具
- 通过依赖于 lambda 的“Boresch”键、角度和扭转约束实现骨架跳跃和绝对结合自由能能力
- 对浮动参考分子支架进行 lambda 依赖的 RMSD 拟合约束
- 非平衡工作框架(Jarzynski 和 Crooks 方程)用于炼金术自由能转换和end-state (e.g., MM->QM or MM->ML) book-ending simulations模拟
- 具有循环闭环和实验约束的蛋白质-配体结合热力学图的网络范围炼金术自由能分析的先进方法
- 使用高斯加速MD(LIG-GaMD)研究配体-蛋白质相互作用的方法。
- 全原子 PME 连续恒定 pH MD(仅限 GPU)。
- 隐式溶剂/显式离子溶剂模型(GBION)。
- 对self-guided朗之万动力学进行更新,使用动量和力引导因素;更新副本交换能力。
以下是Amber24的安装:
安装环境:
Ubuntu 2204,CUDA runtime版本12.4,Cmake 3.22.1,gcc 9.5,g++ 9.5,gfortran 9.5
Amber24 支持的 CUDA 版本: >= 7.5,<12.5。
Amber24 支持的gcc 版本:>= 6, <13
Step 0. 申请下载AmberTools24和Amber24安装包
目前Amber24可以免费申请non-commercial使用,申请地址:Download Amber MD
申请下载两个安装包:AmberTools24.tar.bz2和Amber24.tar.bz2
下载miniforge备用:https://github.com/conda-forge/miniforge/releases/latest/download/Miniforge3-Linux-x86_64.sh
Step 1. 准备工作
sudo apt-get install csh flex gfortran g++ tcsh make gcc flex bison patch bc libbz2-dev wget zlib1g-dev python-tk python3-matplotlib libxml2-dev zlib1g-dev libssl-dev cmake xorg-dev xserver-xorg netcdf-binStep 2. 解压安装包
tar jxvf AmberTools24.tar.bz2
tar jxvf Amber24.tar.bz2Step 3. 编译和安装-CPU串行版
进入到build文件夹,里面有4个文件
cd Amber24_src/build按需要修改其中run_cmake中的安装路径,如果不修改,会安装在Amber24_src旁边。
编译,然后安装。#NOTE: 编译的时候,可能会遇到Miniforge3-Linux-x86_64.sh下载太慢的问题。Ctrl+C退出,将事先下载好的文件复制到 amber24_src/build/CMakeFiles/miniconda/download 目录下,再次运行run_cmake。
./run_cmake
make install -j8设置环境,退出当前Terminal。
source /home/xxxx/amber24/amber.sh如果遇到重新编译,与需要先清除之前的编译残留文件,运行 ./clean_build, 然后再次运行./run_cmake即可。
CPU串行版安装是基本配置,将不会安装支持并行的程序,即.MPI的后缀程序,适用于不强制使用CPU并行的场景。
由于CPU的性能各异,某些并行程序在有些CPU上的并行并不能带来显著的性能提升,还需酌情使用。
Step 4. 编译和安装-CPU并行版
根据需要安装。修改run_cmake中的 -DMPI=TRUE,其他不变,如下
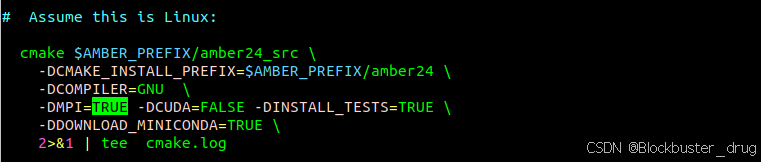
编译,然后安装。
./run_cmake
make install -j8Step 5. 编译和安装-GPU串行版
适用于有1个或多个GPU的情况。
修改run_cmake中的 -DCUDA=TRUE,其他不变,如下
编译,然后安装。
./run_cmake
make install -j8该版本适用于每块GPU单独运行的情况。
在机器上有安装多块GPU时,可以编译GPU并行版本(即调整 -DMPI=TRUE -DCUDA=TRUE ),实际使用中并不能带来显著的提升,不再展示安装效果。
Step 6. 测试安装
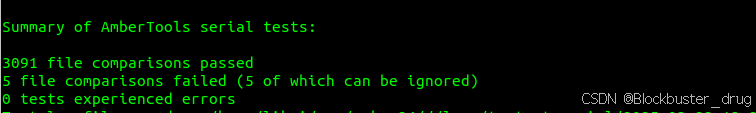
测试CPU串行版:
cd $AMBERHOME
make test.serial结果如下:
测试CPU并行版:
cd $AMBERHOME
export DO_PARALLEL="mpirun -np 4"
make test.parallel结果如下:

测试GPU串行版:
cd $AMBERHOME
make test.cuda.serial结果如下:

对于不通过的情况,找到原因重新安装并测试。