GWAS分析只能基于SNP做?
全基因组关联分析(GWAS)被广泛用于遗传学研究,旨在识别基因型和表型之间的关联。传统的GWAS分析基于SNP位点,且严重依赖单一参考基因组,导致大量存在于不同品种间的结构变异被忽略,而这些变异恰恰是基因多样性的重要来源。
近日,发表在《Genome Biology》的论文,提出了基于k-mer、整合泛参考基因组的GWAS新策略。为小麦白粉病抗性基因的研究提供新的靶点。

研究背景
小麦是全球最重要的粮食作物。白粉菌(Bgt)引起的白粉病是小麦主要病害之一。从古老的地方品种和野生近缘种中发掘新的、持久的抗性基因,成为保障粮食安全的迫切任务。传统的GWAS方法:严重依赖单一参考基因组(如“中国春”),导致大量存在于不同品种间的结构变异。
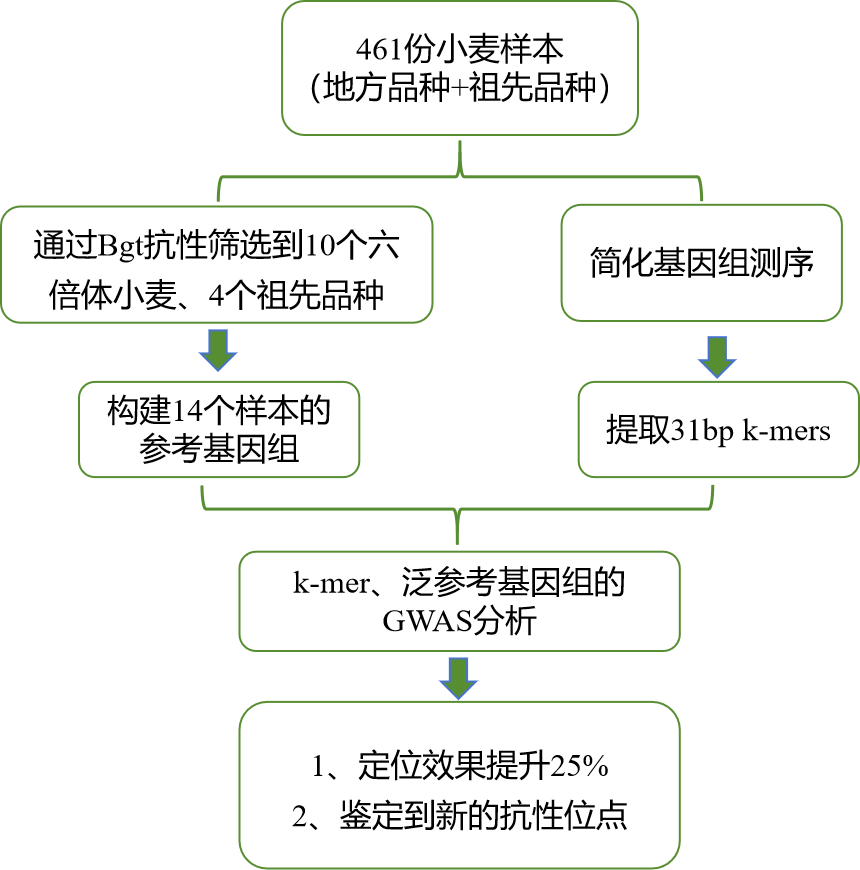
实验设计流程图
主要研究结果
1.发现丰富的抗性基因资源
利用代表全球遗传多样性的10个Bgt菌株,对小麦种质资源进行表型鉴定,发现28份材料对全部10株Bgt表现广谱抗性,暗示可能存在Pm13/Pm36等新型激酶融合蛋白抗性基因。
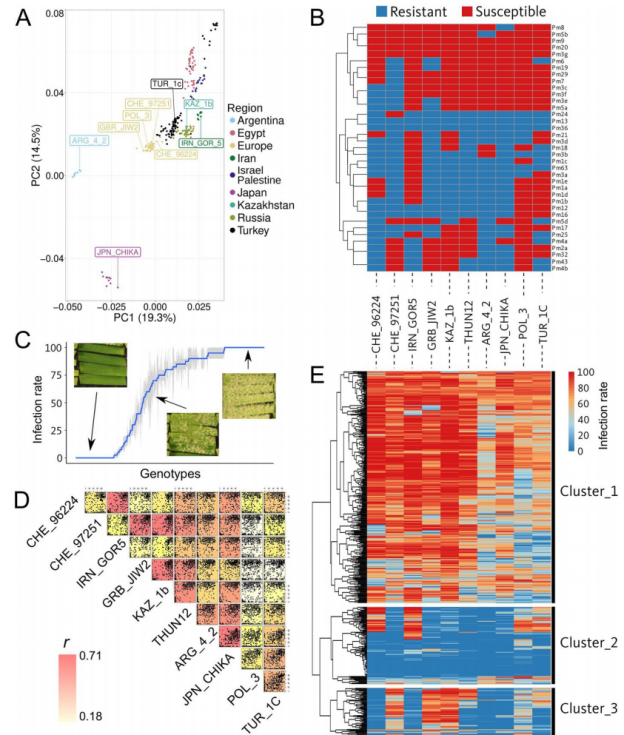
图1 10株小麦具有全球遗传多样性代表的白粉病分离株的表型分析
2.k-mer GWAS显著优势
与芯片法、简化基因组SNP法相比,k-mer方法检出16,895个显著关联k-mer,定位34个抗性位点。 多基因组映射将k-mer定位率从中国春参考基因组的68%提升至93%,新增25%抗性关联k-mer。
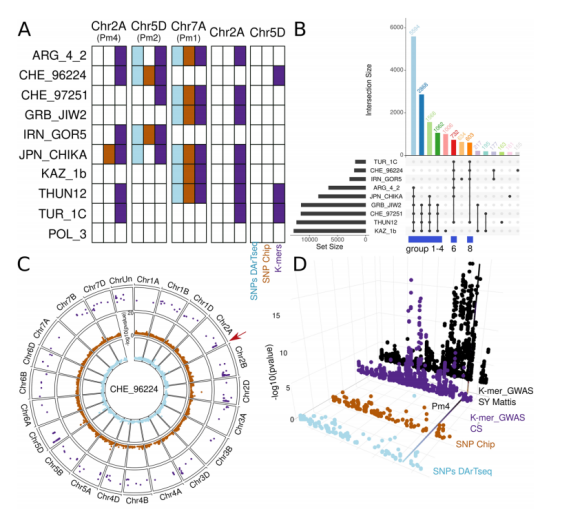
图2 k-mer GWAS结果
3.成株期抗性原理突破
k-mer GWAS方法成功检测到田间成株抗性的多个微效QTL,而传统SNP方法未发现显著信号。
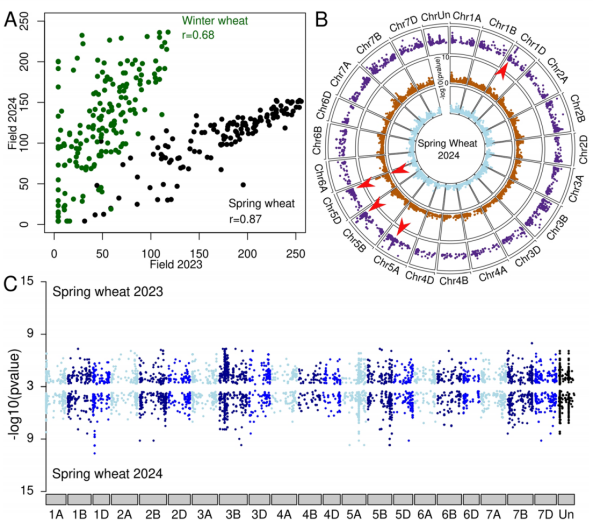
图3 小麦成株期抗性的遗传基础
思考与讨论
本研究首次提出基于k-mer、整合泛参考基因组的GWAS策略,找到了更多与小麦白粉病抗性相关的位点,为GWAS分析提供新的方向。
凌恩 提供动植物基因组重测序、转录组、代谢组等项目,重测序经验丰富,可进行GWAS,mGWAS以及QTL等定位分析,选关键候选基因,解析基因组与各类表型间的关联,开展农作物功能SNP标记“芯片研究以及育种在线分析平台开发,提供专业的种质资源数字化一站式技术服务!