【Linux】snakemake应用之扩增子分析流程
扩增子相关软件安装
新建目录
mkdir 16S_rRNA_amplicon
cd 16S_rRNA_amplicon
创建环境依赖清单文件env.yaml
channels:- conda-forge- bioconda
dependencies:- bioconductor-shortread #R语言中读取DNA序列- bioconductor-dada2- bioconductor-phyloseq- r-pheatmap- r-ggplot2- r-dplyr- r-phangorn- flash #两条reads拼接- cutadapt #裁剪引物- mafft- fasttree- biopython- pandas
创建环境并且安装上述依赖
mamba env create -n 16S_rRNA_amplicon --file env.yaml
# 激活环境
mamba activate 16S_rRNA_amplicon
安装nodejs
#要用ez biocloud进行物种注释
nodejs官网:https://nodejs.org/en/download/
# 下载
wget https://nodejs.org/dist/v20.11.0/node-v20.11.0-linux-x64.tar.xz
# 解压
tar -xf node-v20.11.0-linux-x64.tar.xz
# 删除源文件
node-v20.11.0-linux-x64.tar.xz
# 添加到环境变量
vim ~/.zshrc
export PATH=$PATH:/mnt/d/WSL/software/node-v20.11.0-linux-x64/bin
# 更新
source ~/.zshrc
安装puppeteer
# 创建一个目录
mkdir puppeteer
cd puppeteer
# 初始化工程
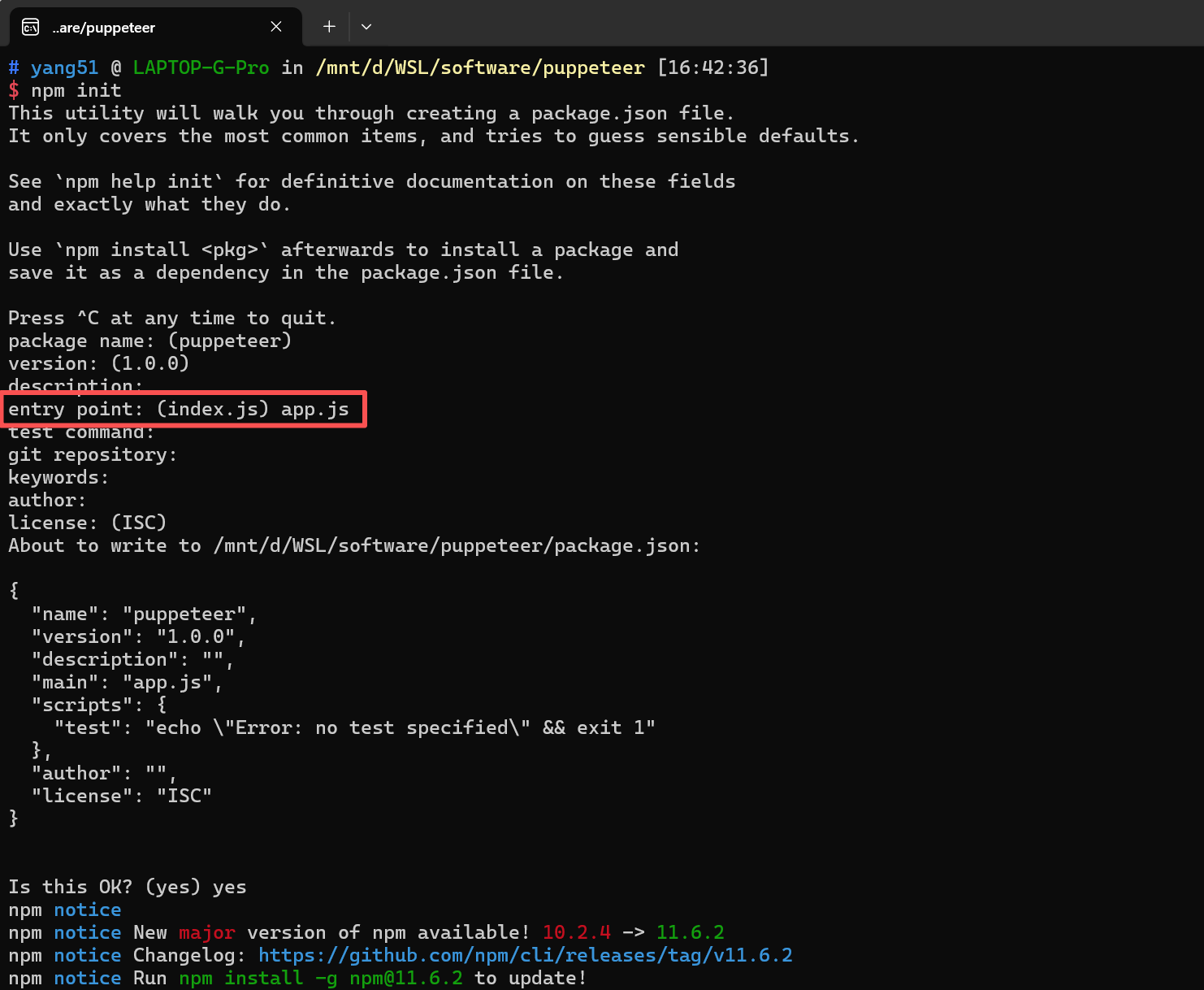
npm init
仅修改entry point,其他默认
# 更改module模式
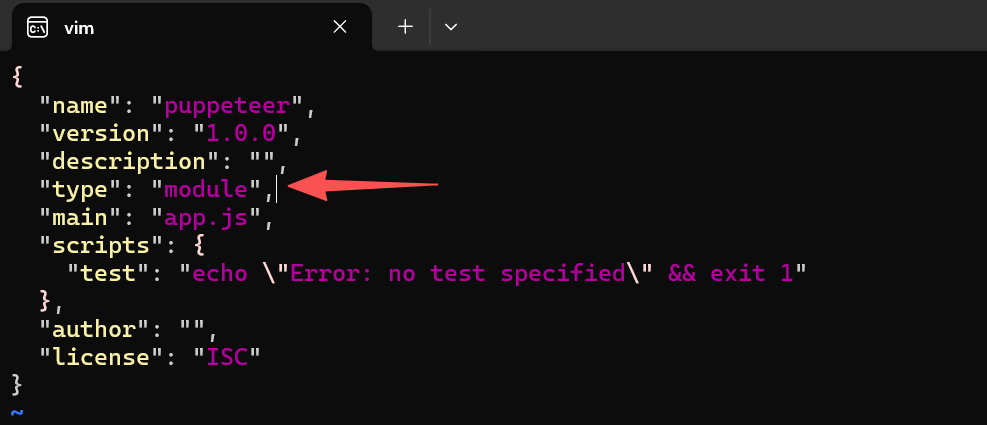
vim package.json
添加一行"type": "module",
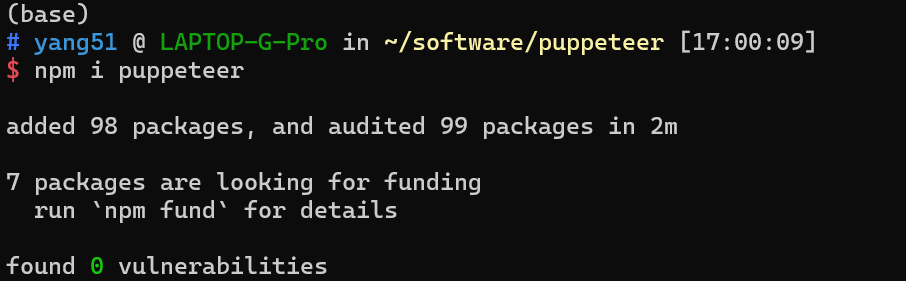
# 安装
npm i puppeteer
注:
该步骤在/mnt/d/WSL/software/puppeteer会报错
移动到/home/yang51/software后方可运行
安装后将app.js文件移动到当前文件夹(流程文件)
分析流程自动化
准备基础工作目录和修改配置文件
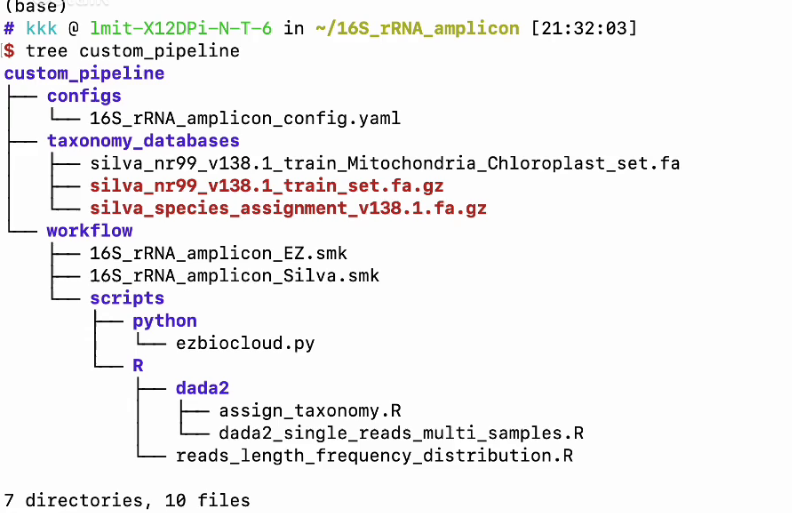
将custom_pipeline.tar.gz放入工作目录D:\WSL\16S_rRNA_amplicon
# 解压
tar -xzf custom_pipeline.tar.gz