苹果公司基于Transformer架构的蛋白质折叠开源模型SimpleFold-安装体验与比较
SimpleFold介绍
github代码![]() https://arxiv.org/abs/2509.18480
https://arxiv.org/abs/2509.18480
arXiv文章![]() https://doi.org/10.48550/arXiv.2509.18480
https://doi.org/10.48550/arXiv.2509.18480
研究背景
已建立的蛋白质折叠模型,如 AlphaFold2和 RoseTTAFold依靠精心设计的架构实现了突破性的精度,这些架构集成了计算量很大的领域特定设计,用于蛋白质折叠任务,例如 AA 序列的多序列比对 (MSA)、成对表示(pair representations)和三角更新(triangle updates)。这些设计选择试图将我们当前对底层结构生成过程的理解硬编码到这些模型中,而不是选择让模型直接从数据中学习,这可能出于多种原因而有益。对于近缘同源物的孤儿蛋白,基于蛋白质语言模型 (PLM) 的方法往往优于依赖 AlphaFold2 等多谱分析 (MSA) 的方法。
尽管折叠模型最初通过重建目标将蛋白质结构预测视为确定性问题,最近的研究探索了构建折叠生成模型。生成方法提供了一种模拟蛋白质结构在自然界中如何呈现的方法,即作为原子系统吉布斯自由能的非确定性最小化器。生成模型自然地捕捉了这种不确定性,并使得生成一系列可行的构象变得简单,而不是单一的确定性输出。基于这一洞见,最近的研究探索了基于扩散和流动的生成模型用于蛋白质折叠,以及从头蛋白质结构生成。
SimpleFold是一种截然不同的架构设计,摒弃了领域特定设计,转而采用一种更为通用的架构设计,该设计已被证明能够有效解决生成式建模问题,并最终能够尽可能高效地利用数据和计算能力。
SimpleFold是一个基于流匹配的折叠模型,它可以直接将蛋白质序列映射到其完整的三维原子结构,而无需依赖多序列比对 (MSA)、成对相互作用图、三角更新或任何其他等变几何模块。架构灵感源自近期基于 Transformer 的文本转图像和文本转三维流匹配模型,重点强调了突破现有架构设计,采用通用的 Transformer 主干网络,并以流匹配训练目标进行端到端训练。本文证明了强大的折叠性能(见图1)无需显式的成对表示、三角形更新或 MSA 即可实现,这显著降低了架构复杂性,并挑战了围绕这些设计必要性的先入为主的观念与之前的蛋白质折叠模型有很大不同,我们总结了我们的贡献如下:
• 重新将蛋白质折叠作为一项条件生成任务,并介绍 SimpleFold,这是一种基于流的变压器折叠模型,可消除 MSA、成对表示和三角模块。
•将 SimpleFold 扩展到 3B 参数,并在约 9M 提取结构和 PDB 实验数据上对其进行训练。
• 最强大的 SimpleFold-3B 模型与采用硬编码启发式设计的折叠基线相比,在折叠方面表现出色,并且在蛋白质集合生成方面也取得了有竞争力的性能。
• 发布了一系列模型,从高效的 100M 模型到大型 3B 模型,以实现最佳性能(见图1 (d))。SimpleFold-100M 恢复∼我们的最佳模型在主要折叠基准测试中的表现达到 90%,即使在消费级设备上推理也非常高效。
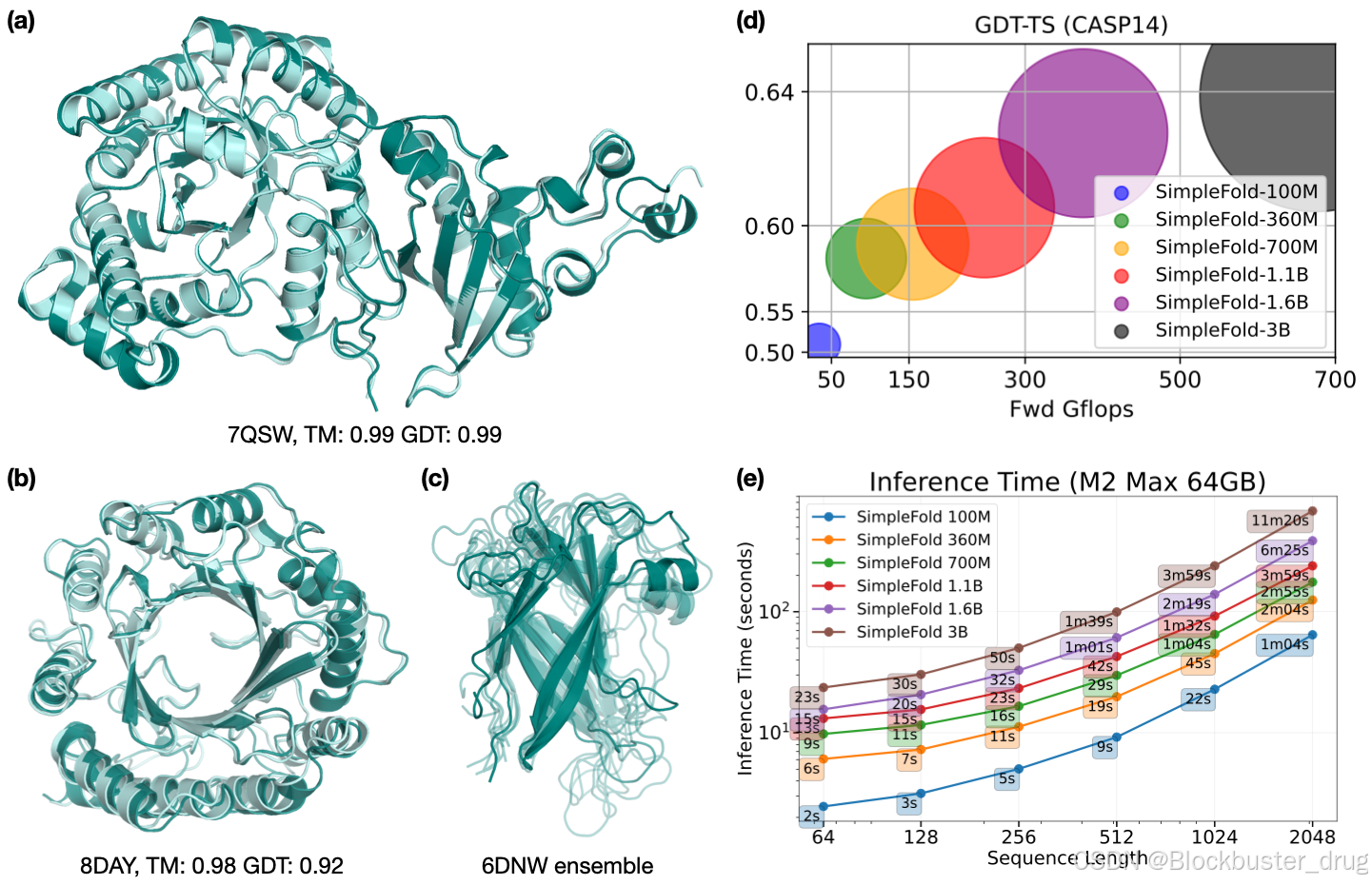
图1:SimpleFold 对目标 (a) 7QSW(RubisCO 大亚基)的 A 链和 (b) 8DAY(二甲基烯丙基色氨酸合酶 1)的 A 链的示例预测,其中浅绿色显示实验结构,深青色显示预测结果。 (c) 使用基于 MD 集合数据微调的 SimpleFold 生成的 6NDW(鞭毛钩蛋白 FlgE)的目标 B 链集合。 (d) 随着模型大小从 100M 增加到 3B,SimpleFold 在 CASP14 上的性能。 (e) 在消费级硬件(即 M2 Max 64GB Macbook Pro)上不同大小的 SimpleFold 的推理时间。
SimpleFold架构
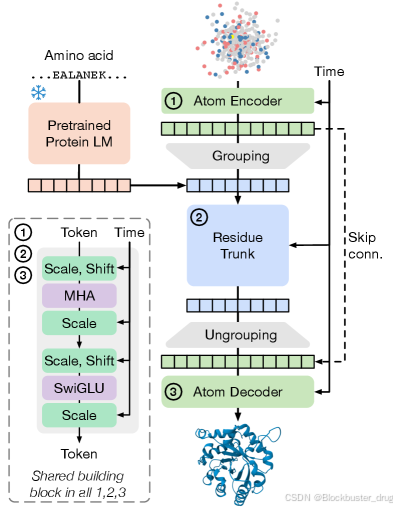
下图SimpleFold架构由三部分组成:轻量原子编码器、重型残基主干、轻量原子解码器
与 AlphaFold2 的架构比较
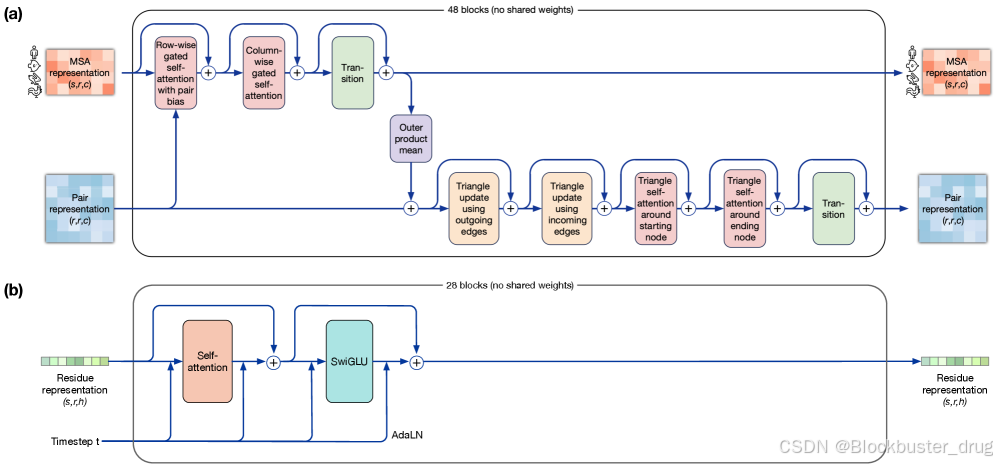
下图展示了 AlphaFold2 和 SimpleFold 中主要计算块的比较借鉴了原始 AlphaFold2 论文。与 AlphaFold 的 Evoformer 模块相比,SimpleFold 遵循简单的 DiT 架构计算效率更高。
实验评估
使用 pLDDT 进行置信度测量
图3 (a) 展示了一个 pLDDT 预测结构的示例,其中红色和橙色表示低 pLDDT,蓝色表示高 pLDDT。如图所示,SimpleFold 对大多数二级结构预测结果有信心,但对柔性环的预测结果不确定。图3 (b) 和 (c) 展示了 pLDDT 与实际 LDDT 的比较。Cα。我们纳入了 CAMEO22 中的目标和 2023 年 1 月之后从 PDB 中随机选择的 1000 条蛋白质链。pLDDT 实现了0.77相对于 LDDT-Cα这表明 SimpleFold 的 pLDDT 模块能够正确地建模预测结构的整体质量。另外需要注意的是,我们的 pLDDT 模块并不遵循生成流程来输出 pLDDT。因此,它可以无缝地应用于衡量其他模型的预测质量,我们将此留待未来研究。
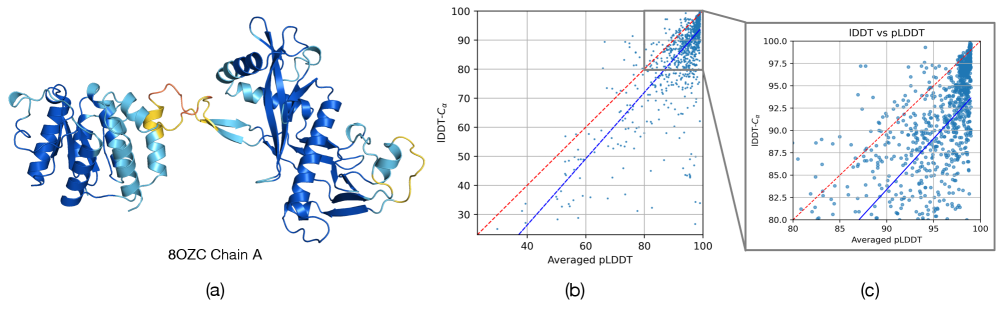
图3:(a)使用 pLDDT 对 SimpleFold 进行预测的示例(颜色从红色到深蓝色表示 pLDDT 从低到高,这是Chakravarty 和 Porter 的可视化结果(2022) ). (b) & (c) pLDDT 和 LDDT 的比较-Cα。
CAMEO22和CASP14蛋白质结构预测基准上评估
为了研究SimpleFold框架在蛋白质折叠任务中的扩展能力,研究人员训练了一系列不同规模的SimpleFold模型(包括100M、360M、700M、1.1B、1.6B和3B)。
研究人员在CAMEO22和CASP14这两个广泛使用的蛋白质结构预测基准上评估了SimpleFold的性能。在CAMEO22上,SimpleFold的表现与目前最先进的模型(如ESMFold、RoseTTAFold2和AlphaFold2)相当。更为重要的是,不使用三角注意力和MSA,SimpleFold在多数指标也能跑到RF2/AF2性能的95%以上。在更具挑战性的CASP14中,SimpleFold甚至超越了ESMFold。SimpleFold跨基准的掉分更小,说明它不靠MSA也能稳健泛化,能够应对更复杂的结构预测任务。

此外,研究还比较了SimpleFold蛋白系综的生成能力。
SimpleFold 模型参数规模的缩放行为
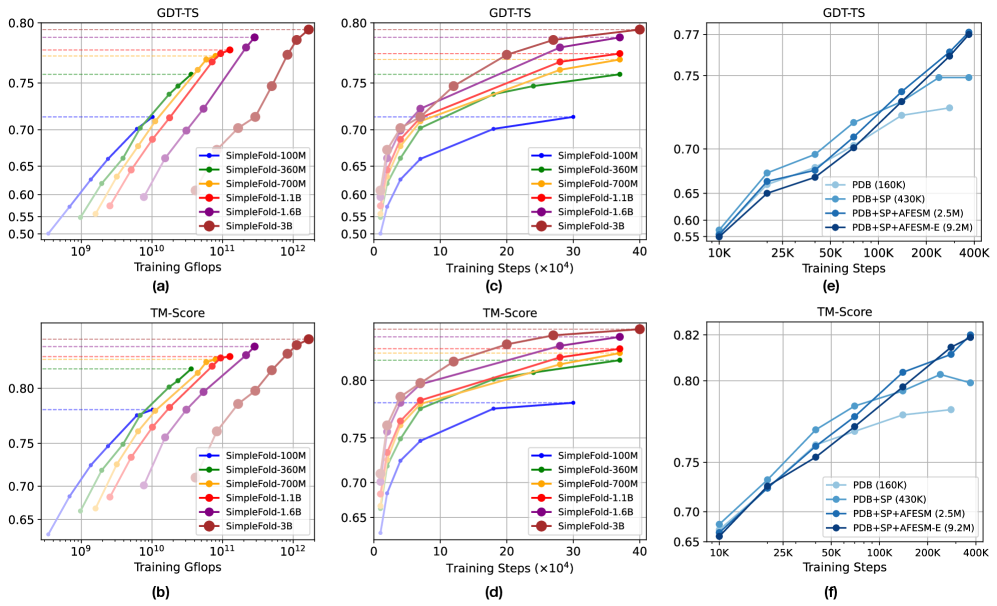
图4:SimpleFold 的缩放行为。训练 Gflops 与 GDT-TS 和 (b) TM-score 上的折叠性能。训练步数与 (c) GDT-TS 和 (d) TM-score 上的折叠性能。数据规模如何影响 (e) GDT-TS 和 (f) TM-score 的性能。所有模型均在 CAMEO22 上进行基准测试。
为了评估在 SimpleFold 中扩大模型规模所带来的效益,我们训练了不同规模的模型,最小的有 1 亿个参数,最大的有 30 亿个参数。所有模型均使用包含 PDB、AFDB 的 SwissProt 和过滤后的 AFESM 的完整预训练数据进行训练。图4 (a)-(d) 展示了模型规模如何影响折叠性能(另见图1 (d))。使用更大的训练预算(即训练 Gflops 和训练迭代次数)训练的较大模型更有利于获得更佳性能。我们相信这些结果凸显了 SimpleFold 积极的扩展行为,并指明了在生物学领域获得更强大生成模型的进步方向。
SimpleFold 受益于模型规模的扩大,这一点已被生成模型在视觉和语言生成等其他领域近期的成功所证明。我们注意到,在蛋白质折叠领域,训练数据和模型规模的缩放效应尚未得到严格的研究。在本节中,我们将从模型和数据两个角度实证展示 SimpleFold 的缩放行为,并强调构建强大的生物生成模型的重要考虑因素。
我们还展示了在 SimpleFold 中扩展训练数据的优势。我们使用不同来源的训练数据训练 SimpleFold-700M:(1) 仅 PDB(16 万个结构),(2) 来自 AFDB 的 PDB 和 SwissProt 组合(SP,27 万个结构),(3) 除 PDB 和 SwissProt 之外,还从 AFESM 中筛选出 190 万个代表性蛋白质,以及 (4) 扩展的 AFESM 集 (AFESM-E),其中包含每个簇中除代表性蛋白质之外的其他蛋白质(总共 860 万个结构)。如图4 (e) 和 (f) 所示,随着我们增加数据组合中独特结构的总数,SimpleFold 的最终性能在 40 万次训练迭代后趋于提高。这些实验结果支持了我们的核心贡献,即构建一个简化且可扩展的折叠模型,该模型受益于不断增长的蛋白质数据总量,这些数据无论是通过实验获得还是从不同模型中提取。
展望
SimpleFold是一个基于流匹配的蛋白质折叠生成模型,它与以往方法的架构设计截然不同。SimpleFold 完全由带有自适应层的通用 Transformer 模块构建,摒弃了 AlphaFold2 引入的诸如代价高昂的对表示和三角更新等启发式设计。SimpleFold 使用简单的流匹配训练目标和额外的 LDDT 损失函数进行训练,而不是使用多个特定于蛋白质的损失函数的组合。
这个简化的框架使我们能够在模型大小和训练数据方面对 SimpleFold 进行大规模训练。我们规模最大模型 SimpleFold-3B 在标准折叠任务上展现出极具竞争力的性能。由于其生成性训练目标,SimpleFold 在多个集成生成任务中展现出非常强劲甚至最先进的结果。SimpleFold 凸显了显著简化蛋白质结构预测架构、减少对计算复杂网络模块依赖的潜力。我们相信 SimpleFold 代表了一种颠覆性的蛋白质折叠方法,它依赖于扩大通用架构块来直接从训练数据中学习底层数据生成过程的对称性。
由于 SimpleFold 是基于标准 Transformer 模块构建的简化架构,因此可以使用常见的微调技术应用于特定蛋白质结构数据以及折叠以外的任务。SimpleFold 还可以直接受益于蒸馏,从而加快推理速度并更高效地部署最大的 SimpleFold-3B 模型。除了大型模型之外,我们还发布了更高效的版本 SimpleFold-100M,它更轻量级,部署速度更快,非常适合推理时间受限的情况。
安装步骤
安装环境:
Ubuntu 22,Python 3.10。
下载安装包,切换到主目录:
git clone https://github.com/apple/ml-simplefold.git
cd ml-simplefold创建conda环境并激活:
conda create -n simplefold python=3.10
conda activate simplefold安装:
python -m pip install -U pip build; pip install -e .安装完成。如果你想在苹果芯片的机器上使用 MLX 后端,需要安装:
pip install mlx==0.28.0
pip install git+https://github.com/facebookresearch/esm.git
结构预测使用体验
运行参数解释如下:
simplefold \--simplefold_model simplefold_100M \ # specify folding model in simplefold_100M/360M/700M/1.1B/1.6B/3B--num_steps 500 --tau 0.01 \ # specify inference setting--nsample_per_protein 1 \ # number of generated conformers per target--plddt \ # output pLDDT--fasta_path [FASTA_PATH] \ # path to the target fasta directory or file--output_dir [OUTPUT_DIR] \ # path to the output directory--backend [mlx, torch] # choose from MLX and PyTorch for inference backend
设定参数,运行一个实例,来自最近公开的PDB数据库中的蛋白,我们取其中的Chain A,包含两个结构域,355个氨基酸残基。
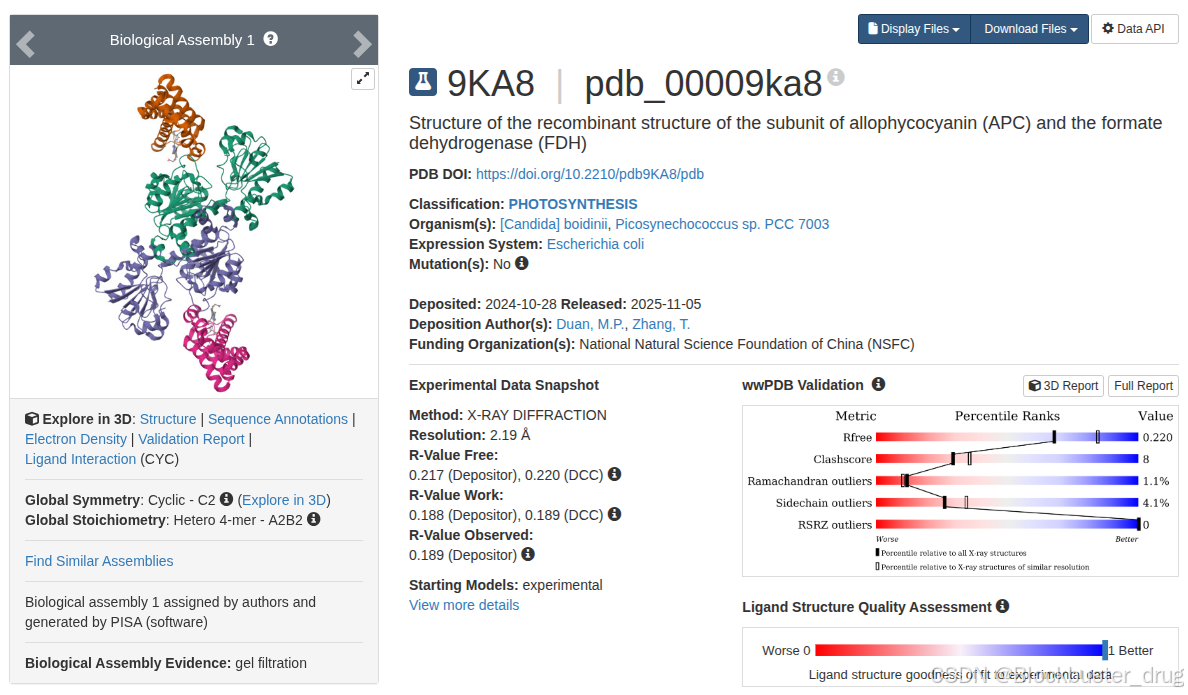
将Chain A的fasta文件保存在主目录下fasta文件夹中,名称:rcsb_pdb_9KA8.fasta,保留3个生成预测结果。在激活的环境中运行:
simplefold \
--simplefold_model simplefold_100M \
--num_steps 500 \
--tau 0.01 \
--nsample_per_protein 3 \
--plddt --fasta_path ./fasta/rcsb_pdb_9KA8.fasta \
--output_dir ./output \
--output_format pdb \
--backend torch运行结果:
初次运行,会下载模型参数文件,保存在cache文件夹下,下载完成后,运行生成预测。结果在output文件夹,目录如下:
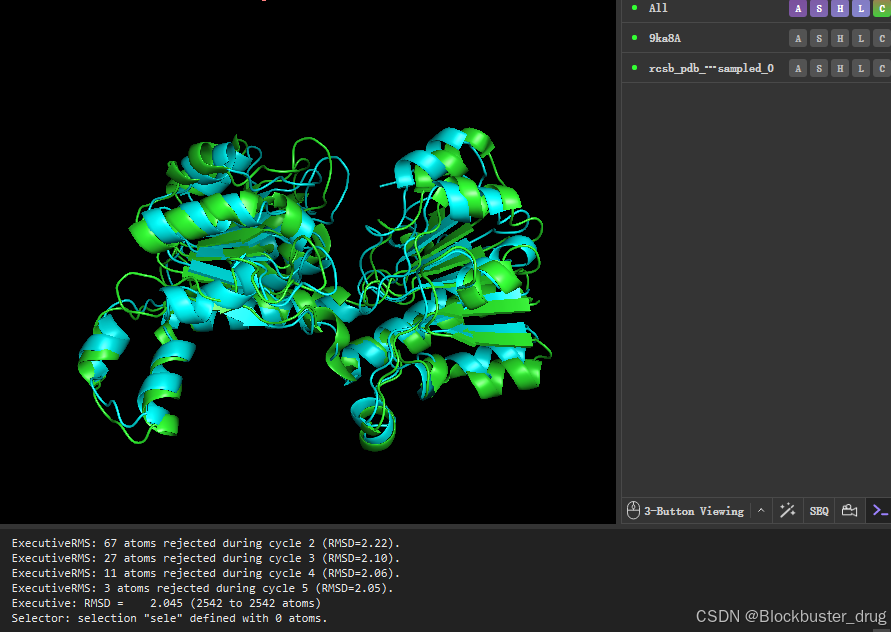
用Pymol打开rcsb_pdb_9KA8_sampled_0.pdb,与PDB结构align之后,RMSD达到2.045,准确度尚可。
参考文献:
1. https://doi.org/10.48550/arXiv.2509.18480