clusterProfile包用于宏基因组学富集分析
https://mp.weixin.qq.com/s/r0xm8SPI4ZjQy46yachXEg 参考
步骤1:获取非冗余基因集(去宿主,组装contigs之后)
# step1 prodigal 预测基因
for i in *_megahit_contigs_rn.fa
donum=${i%%_megahit_contigs_rn.fa}prodigal -a ${num}.faa -d ${num}.fna -p meta -o ${num}.gff -f gff -i ${i}
done# step2 cd-hit 去冗余
cat *.fna > all_cds.fna
cd-hit-est -n 9 -g 1 -c 0.95 -G 0 -M 0 -d 0 -aS 0.9 -T 90 -i all_cds.fna -o all_cds_ref.fna步骤2:获取非冗余基因集的表达量情况(TPM)
# step 1 构建index
~/Software/bwa-mem2/bwa-mem2 index all_cds_ref.fna# step2 bwa-mem2 比对
for i in *_clean_2.fastq.gz
donum=${i%%_clean_2.fastq.gz}~/Software/bwa-mem2/bwa-mem2 mem -t 180 all_cds_ref.fna ${num}_clean_1.fastq.gz ${num}_clean_2.fastq.gz > ${num}_aligen.samsamtools view -Sb -@180 ${num}_aligen.sam > ${num}_aligen.bamsamtools sort -@180 ${num}_aligen.bam -o ${num}_sort.bamsamtools index -@180 ${num}_sort.bamsamtools depth ${num}_sort.bam > ${num}_depth.txtsamtools idxstats -@180 ${num}_sort.bam > ${num}_aligen_result.txtsed -i "1 i GeneID\t\length\tmapped_read\tunmapped_read" ${num}_aligen_result.txt~/Software/my_script/TPM_count.py . ${num}_aligen_result.txt ${num}_TPM.txt# 清理中间文件rm /home/zhongpei/diarrhoea/xjs_FJ_metagenomic/metaMIC_contigs/${num}_aligen.samrm /home/zhongpei/diarrhoea/xjs_FJ_metagenomic/metaMIC_contigs/${num}_aligen.bam#rm /home/zhongpei/diarrhoea/xjs_FJ_metagenomic/metaMIC_contigs/${num}_sort.bamrm /home/zhongpei/diarrhoea/xjs_FJ_metagenomic/metaMIC_contigs/*bam.bairm /home/zhongpei/diarrhoea/xjs_FJ_metagenomic/metaMIC_contigs/${num}_depth.txt
done~/Software/my_script/TPM_together.py --work_path . --file_maker TPM txt --output_name all_100_cds_ref_TPM.txtTPM_count.py
#! /usr/bin/env python
#########################################################
# TPM count
# written by PeiZhong in IFR of CAASimport argparse
import pandas as pdparser = argparse.ArgumentParser(description='TPM count')
parser.add_argument('OperaPath', help='Path that contain your samtools result file')
parser.add_argument('input_name', help='input_name')
parser.add_argument('output_name', help='output_name')args = parser.parse_args()OperaPath = args.OperaPath
input_name = args.input_name
output_name = args.output_nameif OperaPath.endswith("/"):OperaPath = OperaPath
else:OperaPath = OperaPath+"/"
target = OperaPath+input_name# 'TPM count'
#############################################
def TPMcount(target):table1 = pd.read_table(OperaPath+input_name, index_col=0)table1 = table1[table1.index != "*"]table1["Ng/Lg"] = table1['mapped_read'] / table1['length']SUM_NjchuLj = table1["Ng/Lg"].sum()table1["TPM"] = table1['Ng/Lg'] / SUM_NjchuLj * 1e6table1["TPM"] = table1["TPM"].astype(float)result = table1[["Ng/Lg","TPM"]]print(SUM_NjchuLj)result.to_csv(OperaPath+output_name, index=True, sep="\t", float_format="%.6f")
#############################################TPMcount(target)
print(target)TPM_together.py
#! /usr/bin/env python
#########################################################
# TPM count file combine
# written by PeiZhong in IFR of CAASimport argparse
import os
import pandas as pdparser = argparse.ArgumentParser(description='TPM count file combine')
parser.add_argument('--work_path', '-p', help='Path that contain your samtools result file')
parser.add_argument('--file_maker', '-m', nargs='+', help='If there are multiple makers, separate them by spaces')
parser.add_argument('--output_name', '-o', help='output_txt_name')args = parser.parse_args()OperaPath = args.work_path
file_makers = args.file_maker
output_name = args.output_nameos.chdir(OperaPath)
files = os.listdir(OperaPath)
ls = []
for file in files:if all(file_maker in file for file_maker in file_makers):ls.append(file)
ls.sort()
print(ls)all_data = pd.DataFrame()for file_name in ls:df = pd.read_csv(file_name, sep='\t')df['file_name'] = file_nameall_data = pd.concat([all_data, df])pivot_data = all_data.pivot(index='GeneID', columns='file_name', values='TPM')
pivot_data = pivot_data.round(2)
pivot_data.to_csv(f'{output_name}', sep='\t')得到tpm表
步骤3:eggnog对蛋白序列进行注释(快)
python ~/Software/eggnog-mapper/emapper.py -i all_cds_ref.faa -o all_cds_ref_eggnog --cpu 90 --usemem --override --evalue 1e-5eggnog_kegg_TPM.py --result all_cds_ref_eggnog.emapper.annotations --tpm all_100_cds_ref_TPM.txt --out eggnog_kegg_tpm.txteggnog_kegg_TPM.py
#! /usr/bin/env python
#########################################################
# mix eggnog(kegg) result with tpm
# written by PeiZhong in IFR of CAASimport argparse
import pandas as pd# Parse command-line arguments
parser = argparse.ArgumentParser(description='Mix eggnog(kegg) result with TPM')
parser.add_argument('--result', "-r", required=True, help='Path to eggnog result file')
parser.add_argument('--tpm', "-t", required=True, help='Path to TPM table file')
parser.add_argument('--out', "-o", required=True, help='Path to output file')args = parser.parse_args()# Step 1: Read input files
print("Reading input files")# Read dbcan result
df_result = {}
df_kegg = set() # Use a set to store unique CAZy families
with open(args.result, "r") as f:for line in f:if "#" not in line:protein_id = line.split("\t")[0]kegg_str = line.split("\t")[11]if "-" != kegg_str:df_result[protein_id] = kegg_str# Extract CAZy families and remove duplicatesfamilies = set(entry.split(":")[1].strip() for entry in kegg_str.split(','))df_kegg.update(families) # Add unique families to the global set# Read TPM file
df_tpm = pd.read_csv(args.tpm, sep='\t')# Step 2: Process dbcan results and calculate TPM sums for each sample
print("Processing dbcan results and calculating TPM sums for each sample")# Initialize a dictionary to store TPM sums for each CAZy family and sample
kegg_tpm_sums = {ko: {sample: 0.0 for sample in df_tpm.columns[1:]} for ko in df_kegg}# Convert TPM table to a dictionary for faster lookup
tpm_dict = df_tpm.set_index(df_tpm.columns[0]).to_dict(orient='index')# Process each protein in the dbcan result
for protein_id, kegg_str in df_result.items():# Convert protein ID to gene ID by removing trailing "_number"if "_" in protein_id:gene_id = protein_id.rsplit("_", 1)[0] # Split from right on the last "_"else:print(f"Warning: Protein ID {protein_id} has no underscore, using as gene ID")gene_id = protein_id# Get TPM values for this geneif gene_id not in tpm_dict:print(f"Warning: No TPM values found for {gene_id} (protein {protein_id})")continuetpm_values = tpm_dict[gene_id]# Extract unique CAZy families for this proteinfamilies = set(entry.split(':')[1].strip() for entry in kegg_str.split(','))# Update TPM sums for each unique CAZy familyfor family in families:if family in kegg_tpm_sums:for sample in df_tpm.columns[1:]:kegg_tpm_sums[family][sample] += tpm_values[sample]else:# Dynamically add new CAZy familieskegg_tpm_sums[family] = {sample: tpm_values[sample] for sample in df_tpm.columns[1:]}# Create and save output DataFrame
output_df = pd.DataFrame.from_dict(kegg_tpm_sums, orient='index')
output_df.index.name = 'CAZy_Family'
output_df.to_csv(args.out, sep='\t', float_format='%.2f') # Round to 2 decimal places
print(f"Results saved to {args.out}")步骤4:富集分析 R包clusterProfile
准备一个metadata表

准备一个基因丰度信息表
eggnog_kegg_tpm.txt
# 清除全局环境中的所有对象
rm(list = ls())#安装和加载相关R包(需要安装的删除注释标注“#”把安装命令释放出来)
#pkgs <- c("aplot", "CelliD", "clusterProfiler", "DESeq2",
# "enrichplot", "ggfun", "ggrepel",
# "ggsc", "MicrobiotaProcess", "Seurat")
#install.packages("BiocManager")
#BiocManager::install(pkgs)#加载包
library(MicrobiotaProcess)
library(clusterProfiler)
library(ggplot2)
library(enrichplot)setwd("")#####################宏基因组数据差异分析##################
#1、读取宏基因组数据
meta_mg <- read.table("metadata.txt", header = TRUE) #读取样本分组信息
metagenome <- read.table("eggnog_kegg_tpm.txt",row.names = 1,check.name = FALSE)#读取基因丰度信息#2、定义执行差异分析的函数
DA <- function(expr, # 定义名为 DA 的函数,接收多个参数meta, # 样本元数据表abundance = 'Abundance', # 默认指定丰度列的列名为 "Abundance"group_colname = 'Group', # 默认分组列的列名为 "Group"force = TRUE, # 是否强制执行差异分析relative = FALSE, # 是否进行相对丰度转换subset_group, # 子集分组,指定要保留的组diff_group, # 差异组,指定要比较的目标组filter.p = 'pvalue', # 差异分析的 p 值筛选条件...) { # 允许传入其他参数# 构造差异分析的分组列名sign_group_colname <- paste0('Sign_', group_colname)# 将表达数据转换为 MPSE 对象mpse <- MPSE(expr)# 将元数据与 MPSE 对象合并mpse <- mpse |> left_join(meta, by = "Sample")# 筛选样本中属于 subset_group 的组mpse |>dplyr::filter(!!as.symbol(group_colname) %in% subset_group) |># 使用 mp_diff_analysis 函数进行差异分析mp_diff_analysis(.abundance = !!as.symbol(abundance), # 使用指定的丰度列.group = !!as.symbol(group_colname), # 使用指定的分组列force = force, # 是否强制执行分析relative = relative, # 是否相对丰度转换filter.p = filter.p, # 设置 p 值过滤条件... # 传入额外参数) |># 提取差异分析得到的特征mp_extract_feature() |># 筛选出属于 diff_group 的差异特征dplyr::filter(!!as.symbol(sign_group_colname) == diff_group) |># 提取 OTU 信息作为结果dplyr::pull(OTU) |> suppressMessages() # 隐藏冗余消息
}#鉴定差异表达基因
#3、定义一个包含分组信息的列表,键为组名,值为组标记
groups <- c(Healthy = 'Healthy', Diarrhea = 'Diarrhea')
# 使用 lapply 遍历 groups 列表,对每个分组执行 DA 函数
de_gene <- lapply(groups, function(x) {DA(expr = metagenome, # 传入 metagenome 数据作为表达矩阵meta = meta_mg, # 传入元数据表 meta_mgsubset_group = c('Diarrhea', 'Healthy'), # 筛选出当前组(Diarrhea)和对照组(Healthy)diff_group = x # 将当前组(Diarrhea)指定为差异组)
})#差异基因的功能分析
#4、执行基因功能富集分析,并比较不同分组的结果
gene_enrich_result <- compareCluster(geneClusters = de_gene,fun = "enrichKEGG",organism = "ko",pvalueCutoff = 0.05,qvalueCutoff = 0.05
)#5、宏基因组富集结果可视化
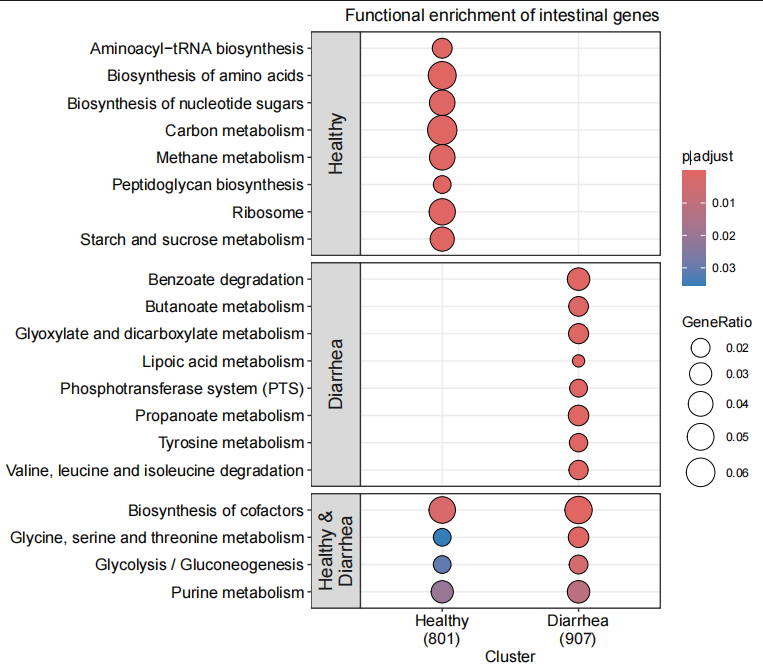
# 使用 dotplot 可视化基因富集分析结果
p1<-dotplot(gene_enrich_result, # 富集分析结果对象facet = "intersect", # 按交集(intersect)分面显示showCategory = 8, # 每组显示前 10 个富集功能split = "intersect", # 根据交集分组label_format = 60 # 标签的最大字符宽度,超出部分换行
) +ggtitle("Functional enrichment of intestinal genes") + # 设置图标题scale_size_continuous(range = c(4, 10)) +theme(plot.title = element_text(hjust = 1) # 调整标题位置,右对齐)
p1
#6、输出结果
ggsave("microbiome genes_all.pdf",plot= p1, height = 7, width =8)
write.csv(gene_enrich_result, file = "metagenome_results_all.csv")