Xenium空间转录组实战 | Xenium Explorer 多样本拼片拆分 ROI 区域圈选
随着 10x Xenium 平台在组织水平上的空间转录组应用越来越广,实验和分析过程中经常会遇到以下两个典型问题:
多样本拼片 (Multi-sample montage)
一张 Xenium 载玻片上往往放置多个组织,最终生成的数据包含多个样本区域。
下游分析前需要 对不同样本进行拆分,避免混淆。
ROI 区域圈选 (Region of Interest selection)
有些研究只聚焦于组织的特定区域(例如肿瘤核心、浸润边界、特定解剖区)。
如果整张组织都进入分析,不仅计算量大,还可能引入背景信号。
这时需要 在 Xenium Explorer 里手动圈选 ROI,只导出目标区域进行分析。
最近有好几个小伙伴遇到了这个问题,今天就和大家一起看看如何使用 Xenium Explorer 来完成这两个任务,并在 Scanpy 中进一步分析。
🔹 Step 1. 打开 Xenium Explorer
安装完成后,双击 .xenium 文件即可打开。软件会自动加载组织影像和空间转录组细胞点位。
🔹 Step 2. 多样本拼片拆分
👉 需求场景:一张 Xenium 载玻片上有多个组织,需要拆分成不同样本。
操作步骤:
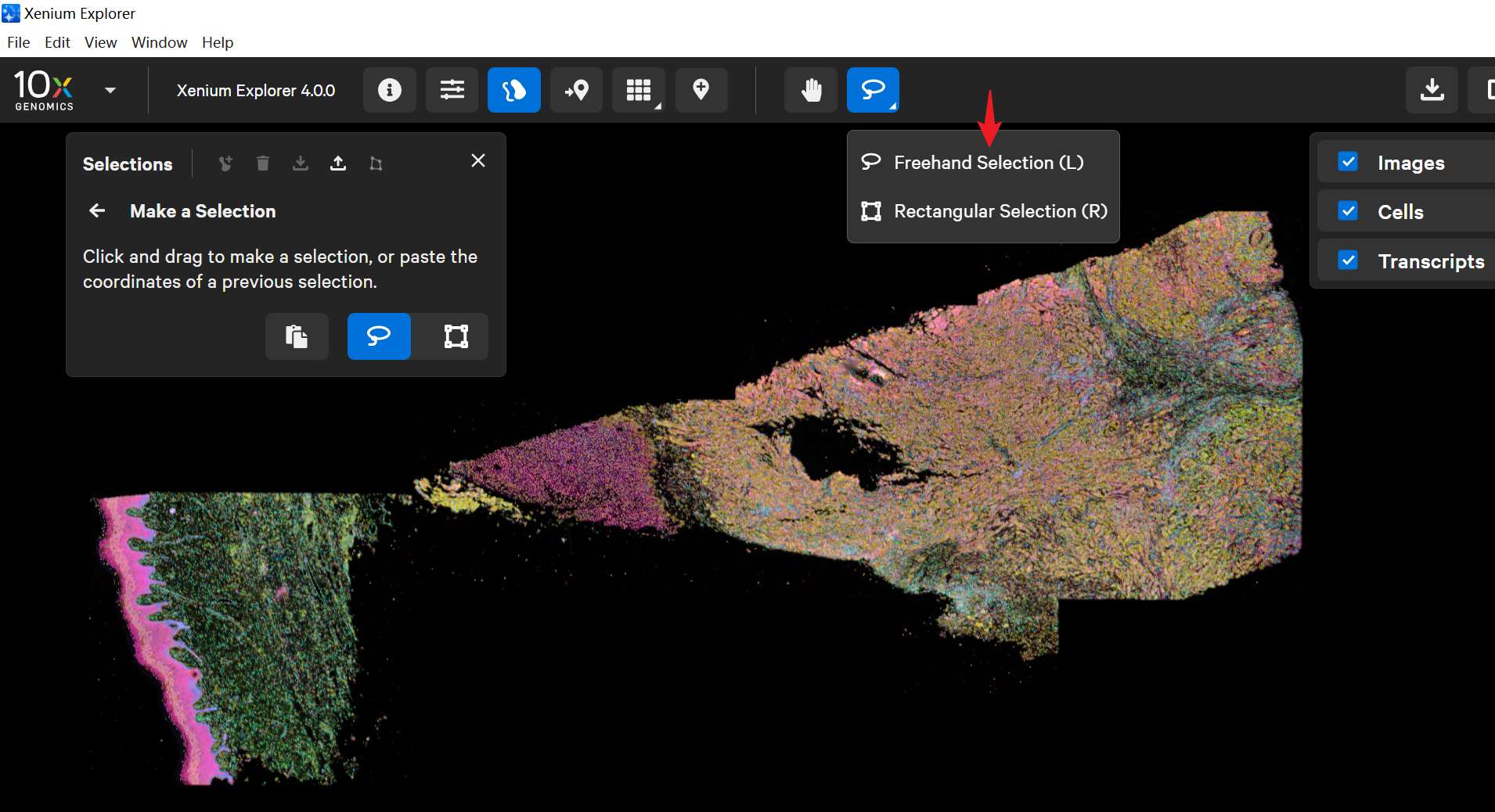
在工具栏中选择 套索 工具。
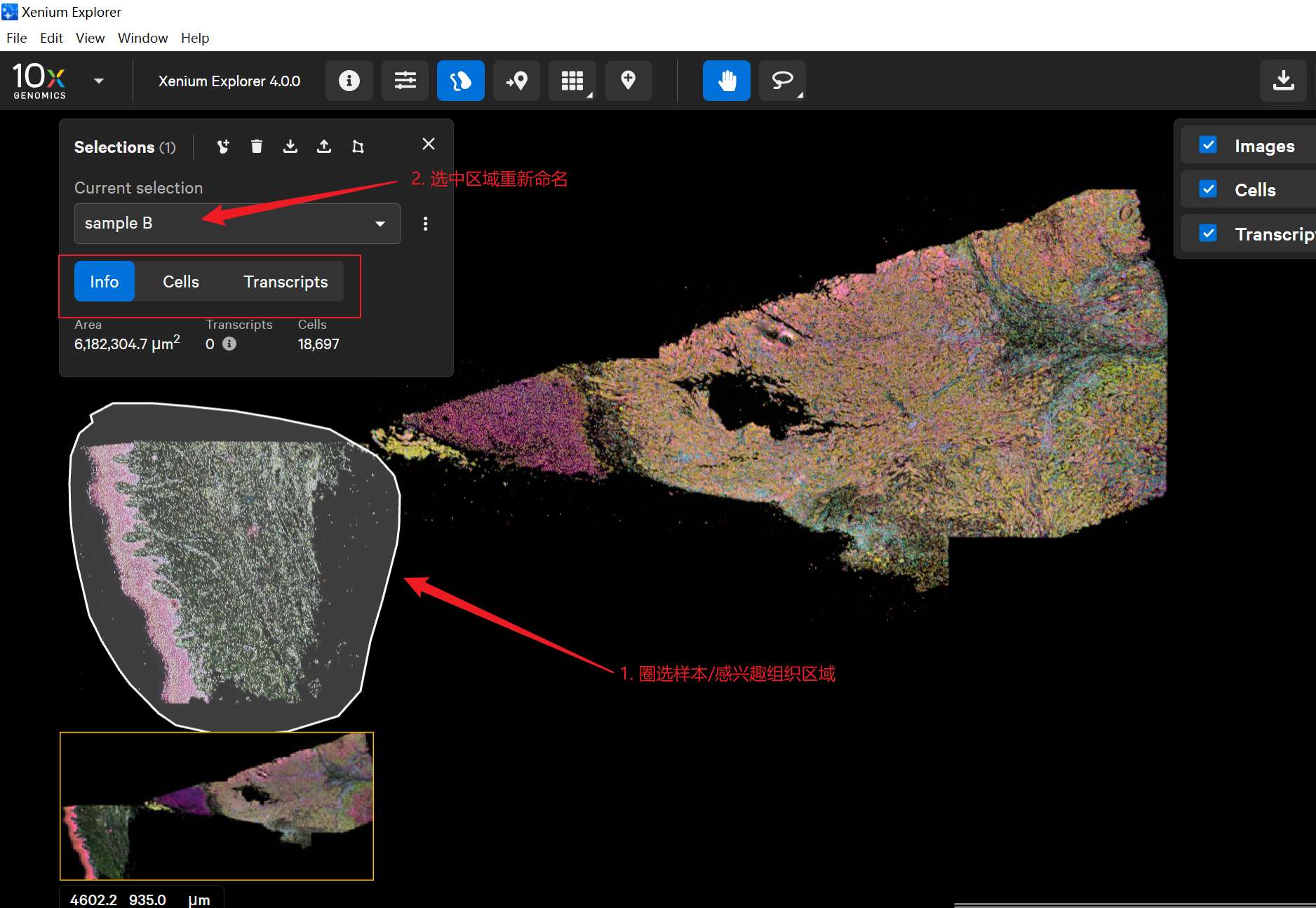
使用套索工具,分别圈选每一个样本区域。
为每个选中组织命名,如
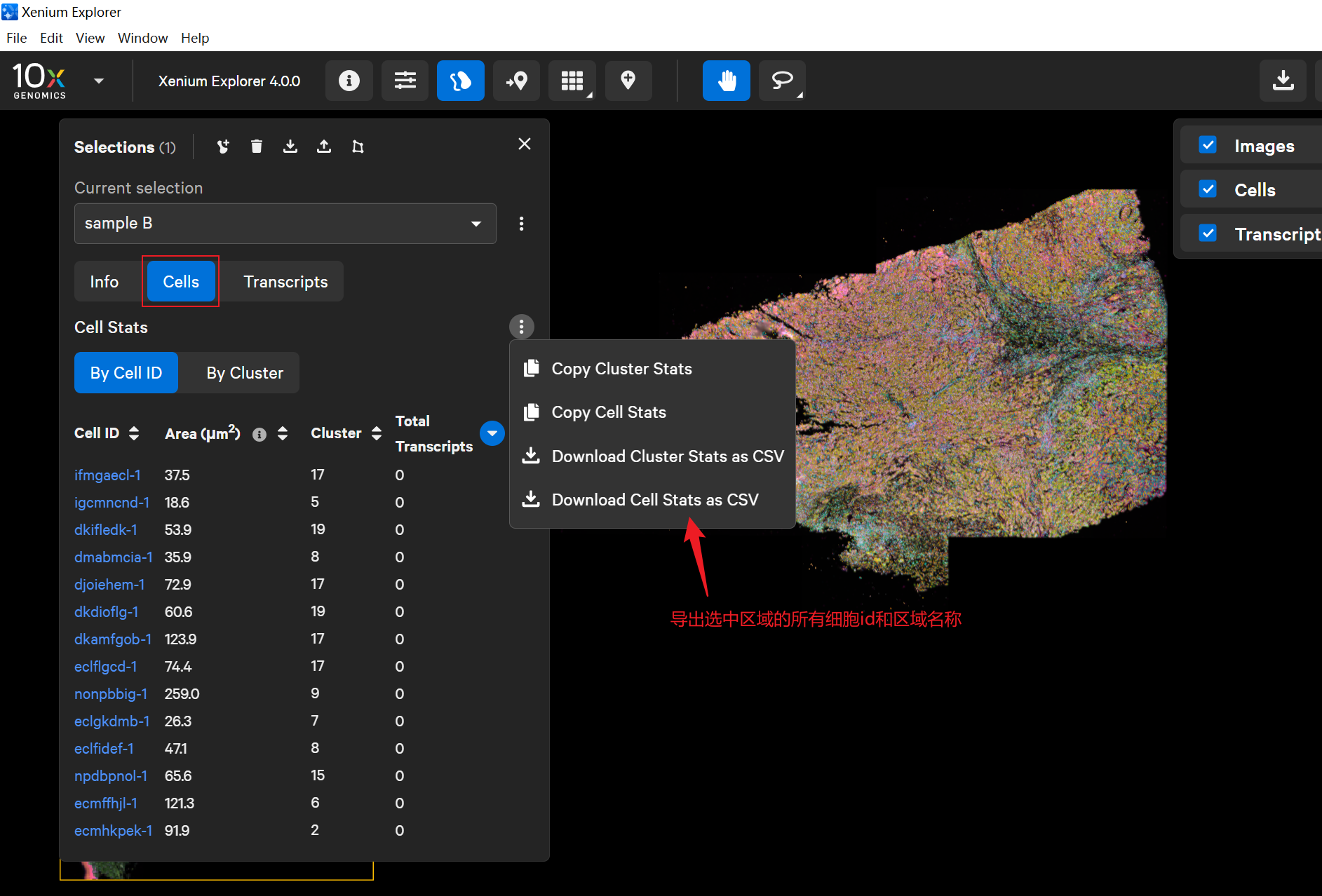
SampleA、SampleB。- 点击 Export → Download Cell Stats as csv。
导出选中区域组织的细胞信息;
每个样本会对应一个单独文件,方便后续分析。
假设切片上是两个样本:
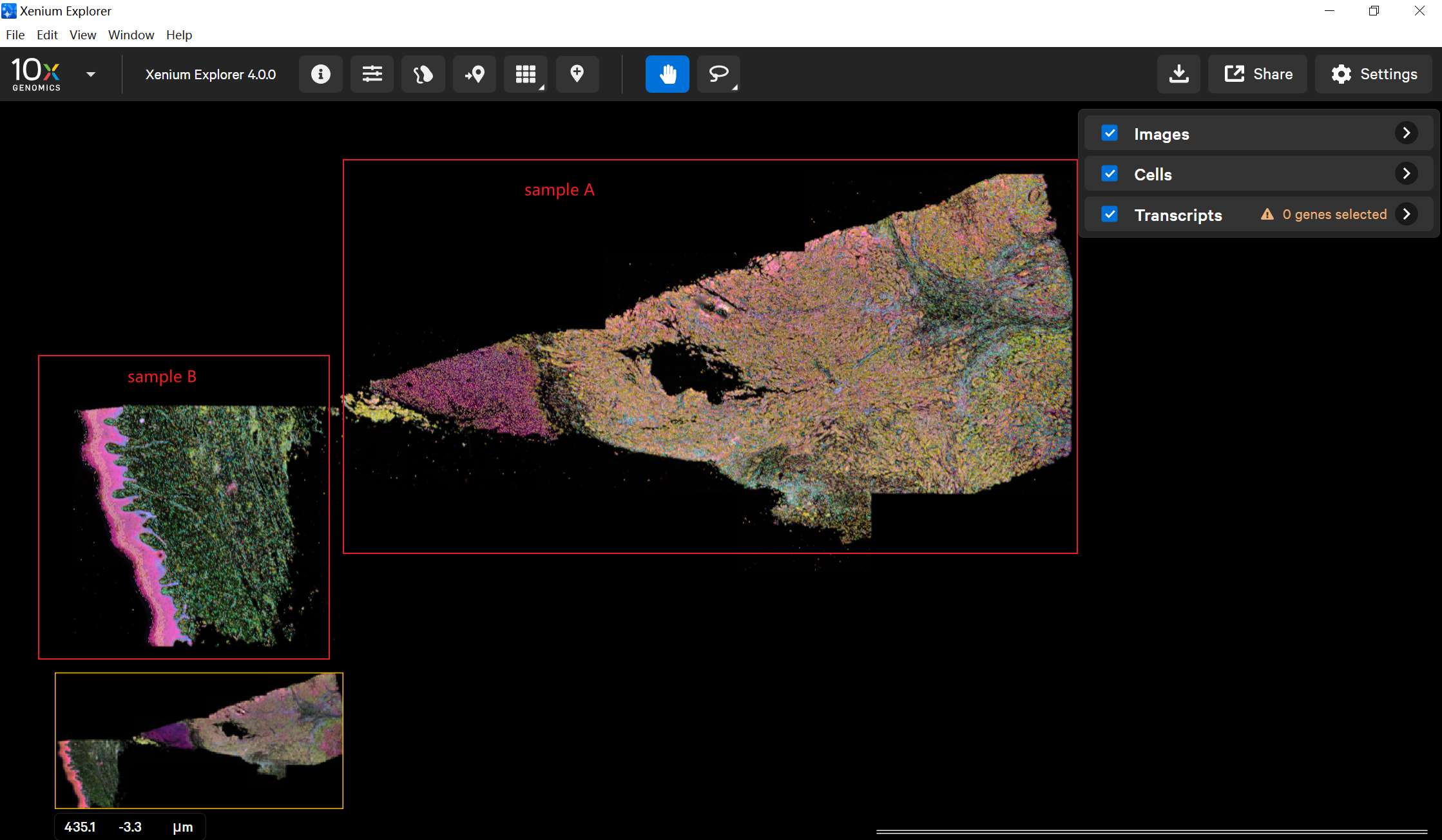
使用套索工具选中组织区域
导出选中样本中包含的细胞信息
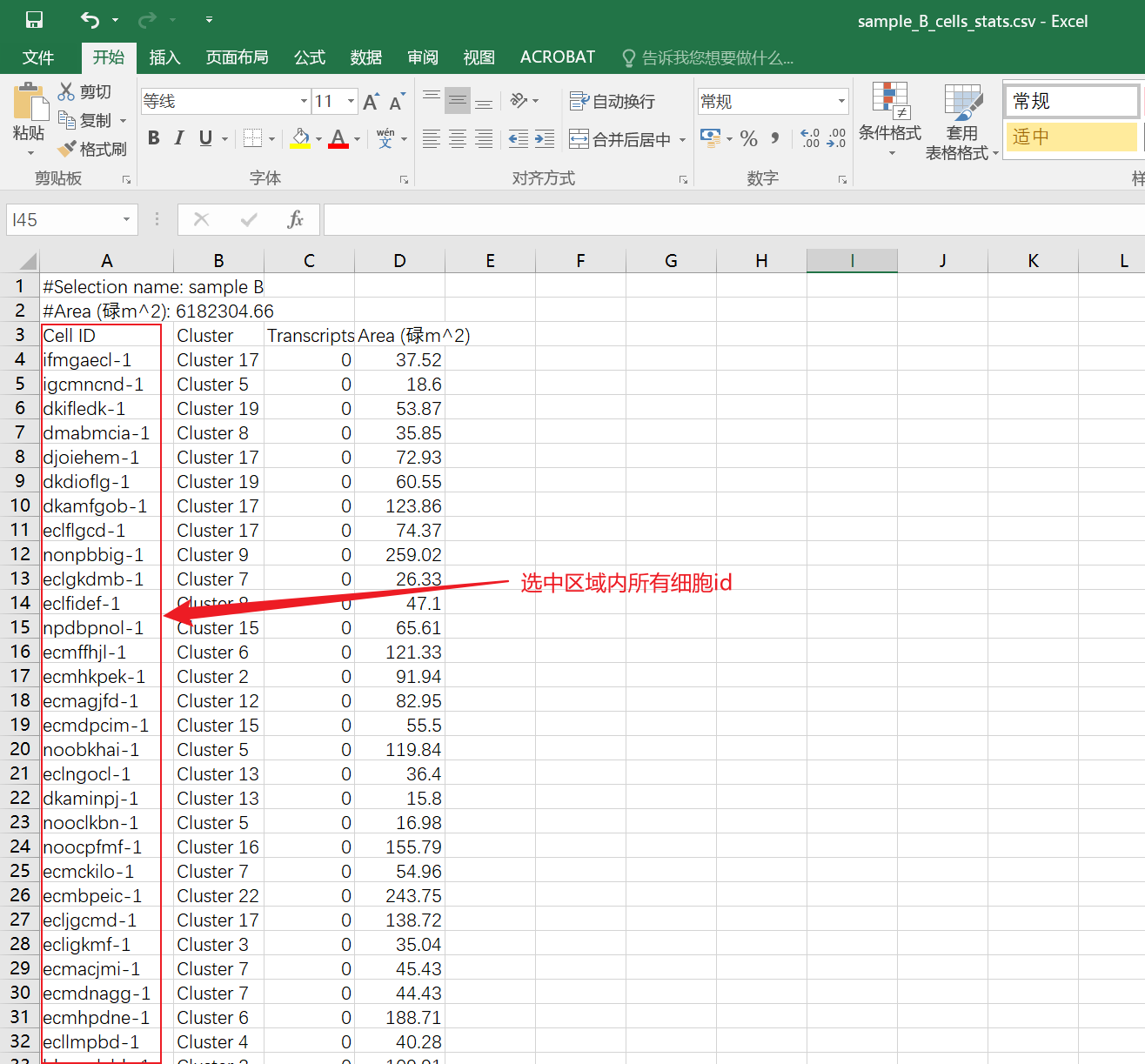
导出细胞id信息
🔹 Step 3. ROI 区域圈选
👉 需求场景:在同一个样本中,只分析局部区域(如肿瘤核心 vs 边缘)。
操作步骤:
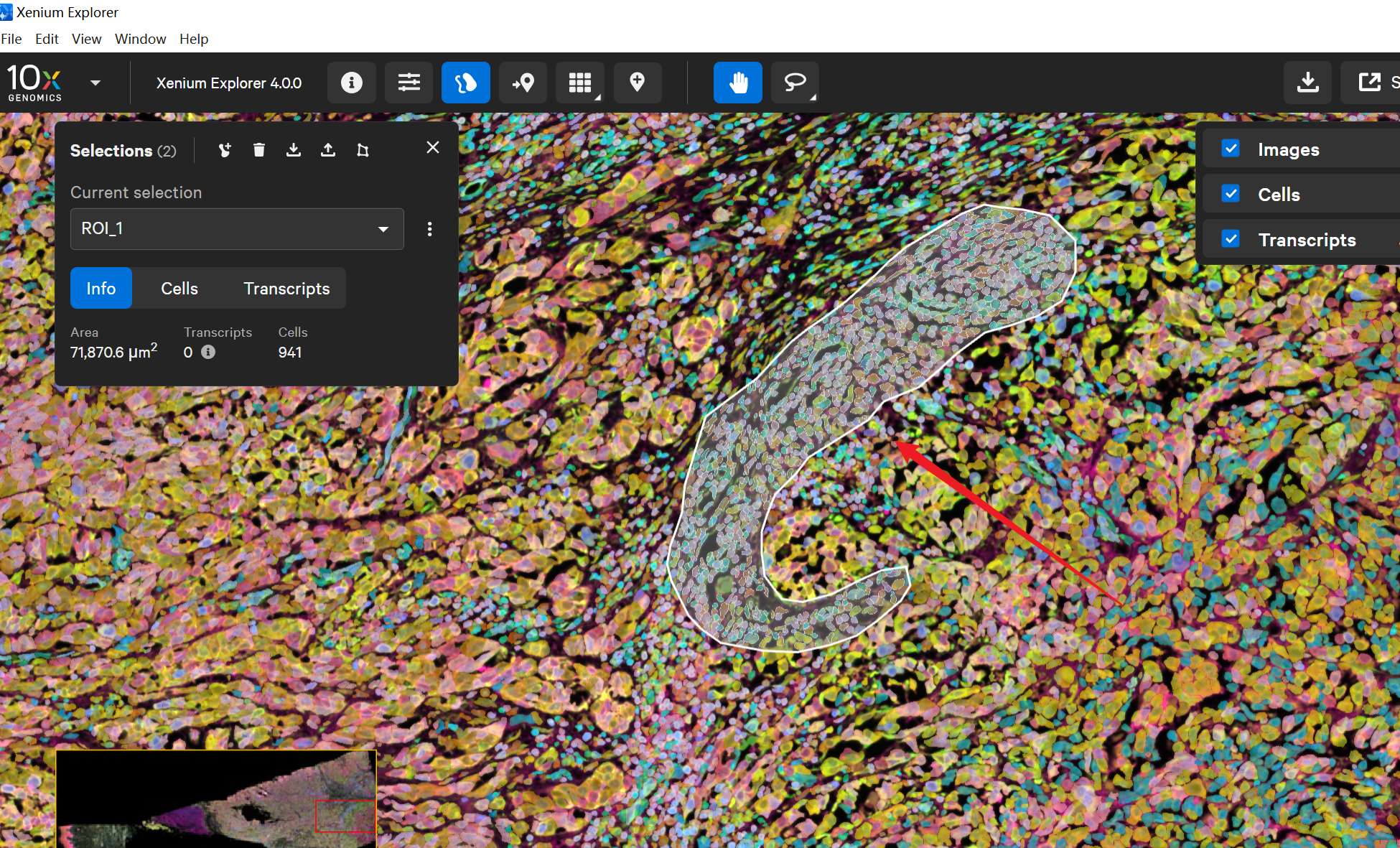
放大至目标区域。
使用套索工具手动圈选目标区域。
如果有多个 ROI,可重复操作,命名为
ROI_TumorCore、ROI_TumorEdge等。导出 ROI 数据,同样选择csv格式。
🔹 Step 4. 在 Scanpy 中加载 ROI 数据
导出的 ROI 文件可直接导入 Scanpy 进行下游分析:
import scanpy as sc
import os# ROI 文件目录
data_dir = "ROI_exports/"
files = [f for f in os.listdir(data_dir) if f.endswith(".csv")]for f in files:df = pd.read_csv(f'./data_dir/f', skiprows=2)df.rename(columns={'Cell ID': 'cell_id'}, inplace=True)df['sample'] = f.replace('.csv', '') # 注意,这里我圈选的区域导出的csv文件是以样本名称命名的tmp.append(df[['cell_id', 'sample']])all_roi = pd.concat(tmp, axis=0)## 然后再把这些信息加入到adata.obs中即可
📌 这样,在 Scanpy 中就能:
分析不同样本之间的差异
比较肿瘤核心 vs 边缘的免疫浸润
构建 ROI 局部的细胞邻近/互作网络
🔹 Step 5. 总结
✨ Xenium Explorer 在空间转录组分析前期非常关键:
多样本拼片拆分 → 保证每个样本独立分析,避免混淆。
ROI 区域圈选 → 聚焦目标区域,提升分析针对性与效率。
📊 在下游 Scanpy 分析中,结合 ROI 标签,可以进行:
差异表达分析
空间邻近分析
细胞类型比较
从而为 肿瘤微环境研究、组织分区比较 等问题提供更精准答案。