【论文阅读】SIMBA: single-cell embedding along with features(2)

代码地址:https://github.com/pinellolab/simba
接上一篇:【论文阅读】SIMBA: single-cell embedding along with features(1)-CSDN博客
摘要
当前大多数单细胞分析流程仅限于细胞嵌入,并且严重依赖聚类方法,而缺乏显式建模不同特征类型之间相互作用的能力。此外,这些方法往往针对特定任务进行定制,因为不同的单细胞问题通常以不同方式被提出。
为了解决这些不足,我们提出了 SIMBA ——一种图嵌入方法,它能够将单细胞及其定义特征(如基因、染色质可及区域和 DNA 序列)共同嵌入到一个共享的潜在空间中。通过利用细胞与特征的协同嵌入,SIMBA 可以支持研究细胞异质性、无聚类的标记物发现、基因调控推断、批次效应去除以及多组学数据整合。
我们展示了 SIMBA 如何提供一个统一的框架,使得多样化的单细胞问题能够在一致的方式下被表述,从而简化新分析方法的开发和向新单细胞模态的扩展。SIMBA 已实现为一个全面的 Python 库(https://simba-bio.readthedocs.io)。
结论
使用 SIMBA 进行单细胞 RNA-seq 分析
scRNA-seq 是用于单细胞分析最广泛的方法。图 2a 提供了 SIMBA 图构建以及在 scRNA-seq 分析中生成的细胞和基因的低维嵌入矩阵的示意概览。SIMBA 将归一化的基因表达矩阵离散化为多个等级(默认五个等级)。随后,通过基因表达水平的加权边将细胞和基因连接起来,构建输入图。接着,SIMBA 通过图嵌入过程生成这些节点的嵌入表示(见图 2a 及方法部分)。根据具体任务,我们可以灵活选择不同层次的可视化,例如展示整个 SIMBA 嵌入(所有细胞与基因的嵌入,补充图 1c)、部分嵌入(仅细胞嵌入,图 2b),或者细胞与可变基因的嵌入(图 2c),乃至任意感兴趣实体的嵌入,并通过 UMAP 进行降维可视化。
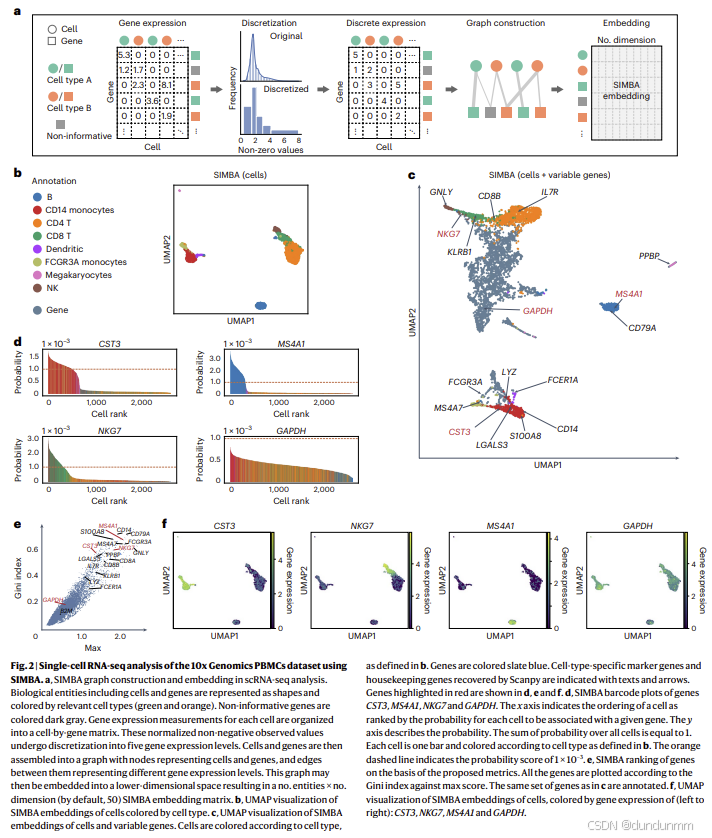
我们将 SIMBA 应用于 10x Genomics 提供的外周血单个核细胞(PBMCs)数据集(补充表 2)。结果显示,SIMBA 的细胞嵌入能够清晰分离八类细胞类型,包括 B 细胞、巨核细胞、CD14 单核细胞、FCGR3A 单核细胞、树突状细胞、NK 细胞、CD4 T 细胞和 CD8 T 细胞(图 2b)。进一步地,细胞与基因的联合嵌入不仅准确恢复了细胞异质性,而且能够将信息性基因嵌入到与其相关的细胞类型附近(图 2c)。先前用于细胞注释的标记基因^2 在 UMAP 图中被高亮显示,证明 SIMBA 不仅能将主要细胞群特异性的基因准确嵌入正确的位置(如 IL7R 嵌入至 CD4 T 细胞,MS4A1 嵌入至 B 细胞),同时也能稳健地识别稀有细胞群的标记基因(如 PPBP 嵌入至巨核细胞),而非信息性管家基因(如 GAPDH 和 B2M)则被嵌入在所有细胞群的中间(图 2c 与补充图 1c)。
这些高亮基因可以通过“条形码图(barcode plots)”进一步验证。条形码图基于恢复的边置信度可视化 SIMBA 对特征分配到细胞的概率估计(图 2d、补充图 1d 与方法部分)。概率分布的不均衡表明某基因与某一细胞亚群(通常对应已知细胞类型)的关联,而概率分布均匀则表示该基因并非细胞类型特异性。对于标记基因(如单核细胞与树突状细胞的 CST3,B 细胞的 MS4A1,以及 NK 和 CD8 T 细胞的 NKG7),我们观察到其在对应细胞类型中的概率明显偏高。相反,对于管家基因 GAPDH,我们观察到其分布更为均匀,且与最高排名细胞的关联概率显著降低。
SIMBA 还提供了一系列定量指标(称为 “SIMBA metrics”),包括最大值、基尼指数、标准差(s.d.)与熵(entropy),可在无需预定义细胞类型的情况下评估特征的细胞类型特异性(方法与补充图 1b、3a)。在“最大值-基尼指数”指标图中(值越高表示细胞类型特异性越强),我们观察到标记基因(如 CST3、NKG7、MS4A1)集中在右上角,而管家基因(如 GAPDH)则位于左下角(图 2e)。所选标记基因的细胞类型特异性进一步通过其在 UMAP 图中的表达模式、SIMBA 条形码图(图 2f,补充图 1d 与 2)以及定量验证(补充说明 2 与补充图 5a)得到证实。SIMBA 指标不仅能够基于细胞类型特异性对特征进行排序,还能过滤掉非信息性特征,从而简化细胞与信息性特征的嵌入可视化,避免 SIMBA 空间因冗余特征而“拥挤”。
我们进一步证明,SIMBA 不依赖可变基因选择,而可变基因选择是 Seurat 或 Scanpy 等标准 scRNA-seq 流程中的关键步骤。当在有或无可变基因选择条件下测试时,SIMBA 均产生了定性相似的嵌入结果(图 2b 与补充图 4e)。不过,我们确实观察到可变基因选择提高了训练效率。我们还将 SIMBA 与依赖聚类的方法^2 以及无聚类方法^6,7 在标记基因检测方面进行了比较(补充说明 3 与补充图 4 和 6)。此外,SIMBA 软件的计算复杂度也与 Scanpy 和 Seurat 进行了基准对比(补充说明 4)。
使用 SIMBA 进行单细胞 ATAC-seq 分析
单细胞 ATAC-seq(scATAC-seq)已被广泛用于描绘染色质开放区域,并识别功能性 顺式调控元件(cis-regulatory elements),例如增强子(enhancers)和活跃启动子(active promoters)。细胞可由不同类型的特征来表征,例如可及染色质区域(“peaks” 或 “bins”),以及包含转录因子(TF)结合基序或 k-mers 的 DNA 序列等顺式调控元件。与现有只能使用 peaks/bins 或 DNA 序列的方法不同,SIMBA 由于其在图构建上的灵活性,可以利用单一或多种类型的特征来学习细胞状态。此外,SIMBA 将“细胞—特征”或“特征—特征”关系以二进制形式(是否存在该特征)编码进图中,因此无需额外的归一化步骤(如大多数 scATAC-seq 分析所需的 TF-IDF)。通过嵌入过程,SIMBA 可同时生成细胞、peaks 和 DNA 序列的嵌入表示。最终,SIMBA 既可以仅可视化部分嵌入(如细胞嵌入),也可以可视化整体嵌入(细胞与所有特征的联合嵌入)。
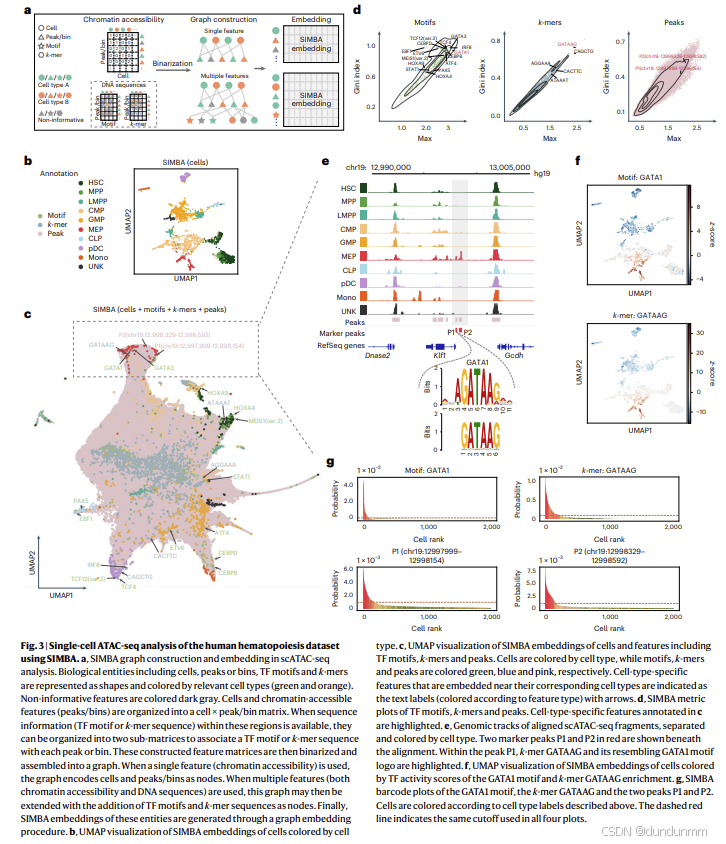
我们将 SIMBA 应用于一个包含 2,034 个经荧光激活细胞分选(FACS)鉴定的人类造血细胞的 scATAC-seq 数据集。结果显示:
-
在仅使用细胞嵌入时,SIMBA 能够准确区分不同类型的细胞,使得基于 FACS 标签定义的不同细胞群体在图中清晰分离。
-
在联合嵌入细胞与多种特征时,SIMBA 成功地将来源于位置(peaks/bins)和序列内容(TF 基序和 k-mers)的不同特征嵌入到同一空间,并依据其生物学关系进行聚合。值得注意的是,根据 SIMBA 的指标,这些嵌入于各细胞类型中的特征均表现出高度的细胞类型特异性。
通过 SIMBA 的分析,我们在人类造血分化过程中得到以下关键发现:
-
SIMBA 识别造血的关键主调控因子(master regulators)
在 UMAP 可视化图中,我们观察到多个已知转录因子的结合基序与相应细胞类型邻近。例如,GATA1 和 GATA3 基序靠近巨核-红系祖细胞(MEP);PAX5 和 EBF1 基序靠近共同淋巴系祖细胞(CLP);CEBPB 和 CEBPD 基序邻近单核细胞(mono)群体。 -
SIMBA 揭示无偏的 DNA 序列(k-mers)并实现基序的“去 novo”发现
我们发现,一些 k-mers 嵌入在其匹配的 TF 结合基序和相关的细胞亚群体附近。例如,DNA 序列 GATAAG 被嵌入于 MEP 细胞,该序列对应 GATA1 的结合基序,而 GATA1 是红系造血的主调控因子。同时,SIMBA 计算的 TF/k-mer 活性评分也显示出 GATA1 基序和 GATAAG 在 MEP 中具有高度活性。 -
SIMBA 识别差异可及的染色质区域,揭示细胞类型特异性调控机制
例如,在 KLF1 基因附近的两个峰(chr19:12997999-12998154 与 chr19:12998329-12998592)主要出现在 MEP 细胞中,其中上游峰(P1)包含 k-mer GATAAG,该序列与 GATA1 结合基序匹配。而 GATA1 已知可调控 KLF1 基因,并在红细胞和巨核细胞的发育中发挥关键作用。因此,SIMBA 通过将这些 MEP 相关调控元件嵌入 MEP 细胞邻域,为研究细胞分化的表观遗传景观提供了新途径。
尽管 SIMBA 在分析策略上有别于传统 scATAC-seq 方法(实现了细胞与特征的联合嵌入),我们仍定性与定量地将其结果与现有先进方法进行了比较,发现 SIMBA 在区分细胞类型的能力上整体优于当前方法。此外,我们验证了在嵌入中加入序列特征对 SIMBA 的结果影响甚微,进一步表明了其在单细胞多组学整合中的广泛适用性。
单细胞多组学分析中的 SIMBA
近年来发展起来的单细胞双组学技术(dual-omics)23–26,能够在同一细胞中同时解析转录组和染色质可及性,从而为探索基因调控规律提供了重要途径。SIMBA 能够从单细胞多组学数据中学习细胞异质性和基因调控网络。图 4a 展示了图构建和 SIMBA 嵌入的过程。研究中将基因表达矩阵与染色质可及性(peaks)、转录因子(TF)基序以及 k-mer 匹配矩阵分别离散化并二值化,然后通过在五类实体(节点)间建立边(cells、genes、peaks、TF motifs、k-mers),构建异质图。随后,图嵌入过程生成细胞与特征的 SIMBA 表征。为了避免信息量不足的 peaks 主导空间(补充图 11a、c),SIMBA 提供了灵活的嵌入可视化,仅呈现部分嵌入结果,以增强细胞及细胞类型特异性特征的可见性。
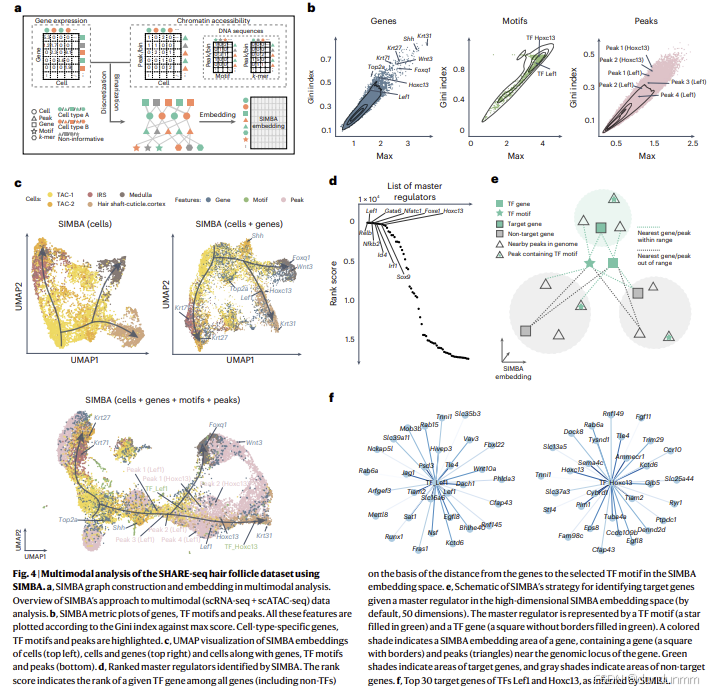
为展示 SIMBA 嵌入的多样性,研究分析了来源于小鼠皮肤 SHARE-seq24 数据中正在经历毛囊分化的细胞群体。首先,研究者计算了 SIMBA 指标(最大值和基尼指数得分),用于评估不同特征类型(基因、TF 基序和 peaks)的细胞类型特异性(见图 4b 和方法部分)。如图 4b 所示,我们成功捕获了与毛囊相关的基因,如 Lef1 和 Hoxc13。同样地,这些基因的邻近区域中 TF 基序和 peaks 也位于指标图的右上象限。
随后,研究者分别可视化和分析了 SIMBA 嵌入的三类对象:(1) 仅细胞;(2) 细胞与基于 SIMBA 指标筛选的高排名基因;(3) 细胞、高排名基因、TF 基序及其邻近 peaks(图 4c)。SIMBA 细胞嵌入结果揭示了从过渡扩增细胞(TACs)分化出的三条命运决策路径,包括内根鞘(IRS)、髓质以及角质层/皮质。进一步结合信息特征的嵌入揭示了毛囊分化轨迹中的关键基因和调控因子。例如,标记基因 Krt71、Krt31 和 Foxq1 分别嵌入到其对应的细胞类型:IRS、角质层/皮质和髓质。而调控因子 Lef1 与 Hoxc13 分别嵌入到角质层/皮质分化的早期和后期阶段。值得注意的是,Lef1 和 Hoxc13 基因座附近的 peaks 也被嵌入到这些基因及基序的邻近区域。TF 基序的距离可能表明转录因子表达与其结合活性的时间差。例如,先锋因子可结合至不可及区域,从而帮助其对其他因子开放。在图 4c 中,Hoxc13 基因的出现早于其基序,这与既有研究结果一致,即 Hoxc13 具有结合不可及基序的能力27。所报道的标记基因和 TF 基序也得到了 UMAP 与 SIMBA 条码图的支持,其预测的细胞类型标签概率很高(补充图 12a–d)。此外,在 SHARE-seq 数据中分别对 scRNA-seq 和 scATAC-seq 单组学数据进行分析,得到的嵌入结果与多组学一致,进一步表明 SIMBA 嵌入方法对输入图中特征的类型与数量均具鲁棒性(补充图 11b、c)。
进一步地,研究表明 SIMBA 提供的细胞与特征共嵌入空间,可用于识别 主调控因子(master regulators) 并推断其靶基因。SIMBA 成功识别了多个先前报道的主调控因子,如 Lef1、Gata6、Nfatc1、Hoxc13,这些因子与小鼠皮肤的谱系承诺密切相关(图 4d、补充表 3、方法部分)。此外,SIMBA 还发现了一个此前未报道的主调控因子 Relb,以及在 TAC-2 细胞群中存在一个新的 Relb+ 细胞亚群(补充图 28、补充说明 6)。
为了推断主调控因子的靶基因,我们假设在 SIMBA 共嵌入空间中:(1) 靶基因同时邻近 TF 基序与 TF 基因;(2) 靶基因座附近的可及区域(peaks)同时邻近 TF 基序与 TF 基因。基于这些 顺式调控动力学假设(图 4e 与方法部分),SIMBA 推断了 Lef1 与 Hoxc13 的靶基因(图 4f、补充图 12e、补充表 4)。值得注意的是,SIMBA 成功恢复了原始研究24 报道的靶基因,包括由 TF Lef1 调控的 Lef1、Jag1、Hoxc13、Gtf2ird1,以及由 TF Hoxc13 调控的 Cybrd1、Hoxc13、St14。
除 SHARE-seq 外,研究还将 SIMBA 应用于另外两个双组学数据集(补充表 2):小鼠大脑皮层的 SNARE-seq 数据23(补充图 13 和 14),以及 10x Genomics PBMCs 的 multiome 数据(补充图 15 和 16)。通过结合细胞类型标签、原始研究中的标记基因以及差异可及的染色质区域对嵌入结果进行验证,进一步证明了 SIMBA 在多组学分析中的适用性。