古多倍体化对被子植物适应性进化的遗传贡献--文献精度154
Genetic Contribution of Paleopolyploidy to Adaptive Evolution in Angiosperms
古多倍体化对被子植物适应性进化的遗传贡献
基于Ks分布的被子植物演化的时间尺度研究--文献精读153-CSDN博客

摘要
古代全基因组重复(WGD 或多倍体化)在植物中广泛存在,且一些 WGD 发生在全球环境剧变的时期。已有研究提出,WGD 可能有助于植物适应环境变化。然而,目前在遗传层面缺乏实证证据来支持这一假设。在本研究中,我们调查了来自多个古代 WGD 事件的基因重复存活者,重点研究了被子植物系统发育树上主要分支中的重复基因,并旨在探索支持多倍体化重要性的遗传证据。我们研究了来自三轮独立 WGD(约 1.2 亿年前 [Ma]、约 6600 万年和 <2000 万年前)事件的重复基因,这些基因在 25 种选择物种中共同保留。在白垩纪-古新世边界时,全球变冷和黑暗是主要的环境压力,许多谱系中在八次独立发生的 WGD 后,涉及低温和黑暗的基因家族常常保留了基因重复。此外,常保留的重复基因可能是贡献于关键胁迫相关通路强韧性的关键因素。进一步研究发现,参与胁迫反应的全基因组转录因子(TFs)往往在 WGD 波次后保留重复基因,而在许多谱系中共同选择的基因重复可能在严重的环境胁迫中发挥关键作用。综合来看,这些结果为古多倍体化在被子植物进化历史中的全球环境变化适应贡献提供了新的见解。
引言
被子植物(或开花植物)是植物界中最具多样性和数量最多的类群,目前已知约有 35 万个物种。查尔斯·达尔文曾描述被子植物从白垩纪中期到晚期的迅速崛起和早期多样化为“一个可憎的谜团”(Friedman,2009)。目前,被子植物构成了地球表面主要的植物群落,覆盖从热带到极地的陆地区域以及水生栖息地。其成功的原因被推测部分与被子植物进化历史中普遍发生的全基因组复制(WGD)事件有关(Levin,1983;Soltis 等,2009;Van de Peer 等,2009;Van de Peer 等,2017)。WGD 长期以来被认为是物种形成、适应和多样化的重要进化力量(Wood 等,2009;Soltis 和 Soltis,2016)。
在过去二十年中,研究者们已经证明,WGD 在开花植物的进化历史中比之前认为的要普遍得多(Bowers 等,2003;Blanc 和 Wolfe,2004;Cui 等,2006;Soltis 等,2009;Jiao 等,2011;Renny-Byfield 和 Wendel,2014;Van de Peer 等,2017)。在现存的被子植物和种子植物多样化之前,已经识别出了两次祖先性的 WGD(Jiao 等,2011)。被子植物中的两个主要类群——真双子叶植物和单子叶植物——都在其进化历史的早期经历了古多倍体化事件,分别被命名为 γ(γ)和 τ(τ)(Jaillon 等,2007;Tang 等,2010;Jiao 等,2012;Jiao 等,2014;Vekemans 等,2012;Ming 等,2015)。此外,WGD 事件还发生在许多物种丰富的群体的共同祖先中,如菊科、十字花科、葫芦科、豆科和禾本科(Cannon 等,2015;Edger 等,2015;Huang 等,2016;McKain 等,2016;Ren 等,2018;Wang 等,2018)。特别地,WGD 在许多谱系中反复发生。例如,拟南芥在与单子叶植物分化后,其进化历史中发生了三轮 WGD(γ-β-α)(Bowers 等,2003);香蕉属(Musa)的谱系在与禾本科分化后也独立经历了三轮 WGD(D'Hont 等,2012)。
此外,先前的研究发现,WGD 的时间分布并非随机分布在被子植物的系统发育树上,这表明 WGD 可能在环境选择中发挥了作用。古老的 WGD 波次在白垩纪-古新世(K-Pg)边界时期独立地出现在许多植物谱系中,暗示 WGD 可能帮助物种度过了这一灭绝事件(Fawcett 等,2009;Vanneste 等,2014)。还有研究提出,多倍体化与晚中新世的 C4 草原扩展以及与最近的冰川最大期的适应有关(Estep 等,2014;Novikova 等,2018)。因此,WGD 被推测与灭绝事件以及其他极端环境变化相关。然而,目前在遗传层面上,关于 WGD 对适应的贡献和意义的证据仍然未被充分探索。
众所周知,多倍体化通过添加一套额外的基因组,同时复制成千上万的基因,为进化提供了大量的原始遗传材料(Adams 和 Wendel,2005;Doyle 等,2008;Hegarty 和 Hiscock,2008;Soltis 等,2015;Van de Peer 等,2017)。在随后的分割和二倍体化过程中,绝大多数基因会迅速恢复为单拷贝状态(Lynch 和 Conery,2000),而保留下来的基因则通过新功能化和亚功能化对遗传创新起着特别重要的作用(Ohno,1970;Force 等,1999)。此外,重复的基因可能还会导致基因调控网络(GRNs)发生变化(Conant,2010;De Smet 和 Van de Peer,2012),这可能有助于植物适应。
为了探索 WGD 的重要性,我们在此全面追踪了 25 个植物基因组中的全球基因家族的进化历史,并调查了独立 WGD 后的遗传修改。首先,使用代表被子植物主要谱系的 25 个已测序植物基因组(补充表1)重建了全球基因家族,并进行了系统基因组分析,以识别在古代 WGD 后保留重复基因的基因家族。然后,我们识别了在经历极端环境变化的某些时期,经过独立 WGD 后保留重复的基因家族,寻找遗传层面的潜在选择信号。最后,通过重建来自 RNA 测序(RNA-seq)数据的 GRN 并整合先前已知的通路,我们提供了证据,显示保留的重复基因如何有助于在应对环境胁迫时重塑 GRN。
结果
WGD 后保留的同源基因的识别
为了识别 WGD 的遗传贡献,我们调查了被子植物进化历史中 21 次公认的多倍体化事件(图1)。我们选择了 25 个已测序的植物基因组(补充表1),并从其蛋白编码序列中构建了假定的基因家族。总共有 66,509 个同源基因组通过 OrthoMCL(Li 等,2003)构建。其中,12,077 个同源基因组含有四个或更多的基因,并包括至少一个来自外群的基因(如藓类植物Physcomitrella patens,种子植物Selaginella moellendorffii,和Amborella trichopoda),这些基因家族用于重建最大似然系统发育树(见方法)。然后,进行了系统基因组分析,以从每个基因家族的系统发育中寻找重复事件,如先前所述(Jiao 等,2011)。
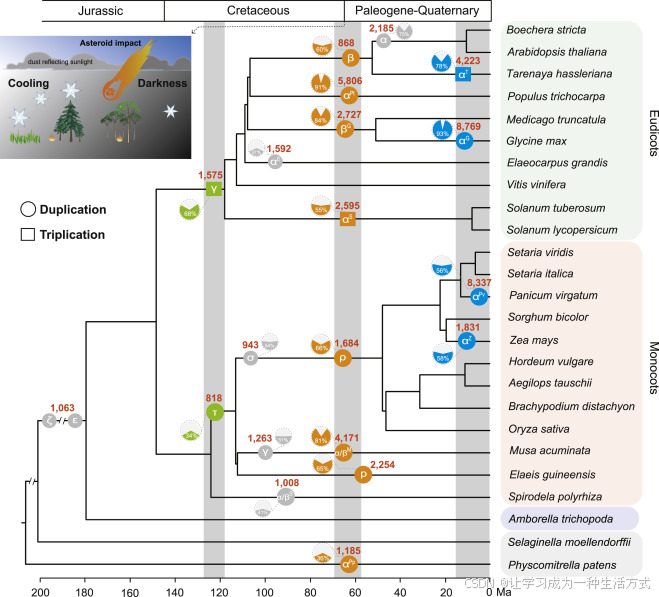
图1. 推测陆地植物进化历史中每次 WGD 后存活的基因家族数量
系统发育树展示了本研究中25种植物的拓扑结构和分歧时间。这25种物种的进化关系基于当前公认的拓扑结构(Angiosperm Phylogeny Website)。每个物种树节点的分歧时间来自 TimeTree 网站(http://timetree.org/)。公认的全基因组重复(圆形)和三倍体(方形)事件被标记在系统发育树的分支上。识别出了三个具有丰富 WGD 的时期(约 120 Ma、约 66 Ma 和 <20 Ma),分别用绿色、橙色和蓝色表示。每次 WGD 后保留重复基因的基因家族数量显示在相应的圆形或方形周围。由同源性证据验证的重复基因在 WGD 后生成的比例,显示在虚线圆圈中。左上方的草图地图显示了白垩纪-古近纪灭绝期的主要环境胁迫。
为了尽可能确保重复基因来自 WGDs,首先根据重复基因的染色体位置移除了串联重复基因,这可以筛选出一些古老的小规模重复(见方法)。此外,每个物种的同源性分析能够为本研究中识别的大部分重复基因提供同源性支持(图1和补充图1)。另外,一些重复难以分类,因为缺乏在两个连续 WGD 事件之间的分支物种,例如 Poales 中的 ρ 或 σ 事件,和 Musa acuminata 中的 α/βM 或 γM 事件。我们采用了同义替代/同义位点(KS)方法来区分来自特定重复事件的基因存活者(见方法)。综合考虑后,我们能够收集到每次 WGD 事件后存活的基因家族(图1)。
WGD 后三个时期的基因保留模式
WGDs 可以在一次事件中生成大量的重复基因,为进化提供了大量的原始遗传材料。如果独立的 WGDs 帮助物种适应环境变化,那么相似功能的基因重复可能会在这些不同的物种中保留下来,因为它们可能已被共同的环境压力所选择。
先前已识别和标定的21次 WGD 被标注在选择的陆生植物物种树上,其中 14 次可以根据发生时间分为三个波次(图1 和 补充表2)。最古老的波次大约发生在 120 百万年前(Ma),当时 γ(Jaillon 等,2007;Jiao 等,2012;Vekemans 等,2012)和 τ(Jiao 等,2014;Ming 等,2015)事件分别发生在真双子叶植物和单子叶植物的早期进化历史中。第二个波次是白垩纪-古近纪边界周围的波次,当时发生了大量 WGD 事件(Paterson 等,2004;Tuskan 等,2006;Rensing 等,2008;Fawcett 等,2009;Schmutz 等,2010;D'Hont 等,2012;番茄基因组联盟,2012;Singh 等,2013;Vanneste 等,2014)。最近的波次发生在 20 Ma 以内,在大豆、苏丹草、哈斯尔塔兰和玉米的进化历史中发生了四次独立的 WGD(Blanc 和 Wolfe,2004;Schmutz 等,2010;Cheng 等,2013;Lu 等,2013)。
我们发现,确实有一些基因家族在许多物种中保留了来自多个独立 WGD 的基因重复(图2A),这些可能是来自特定胁迫环境的选择信号。66 个基因家族在三个时期中共同保留了基因重复(补充图2),这些基因家族主要是蛋白激酶、转运蛋白和蛋白结合基因家族(补充表3)。320 个基因家族保留了来自最古老波次(γ 和 τ)WGD 的重复,这些基因主要与水分缺乏和盐胁迫响应相关(图2B)。这些存活者可能至少部分是由大约 120 Ma 白垩纪时期的干旱气候所选择的(Heimhofer 等,2005)。第二波 WGD 发生在白垩纪-古近纪边界,伴随着严重的环境变化,包括全球变冷、黑暗、酸雨和野火(Nichols 和 Johnson,2008;Schulte 等,2010)。493 个基因家族保留了至少六次独立的 WGD 重复(图2A),这些基因家族富集了许多与胁迫相关的基因本体论(GO)术语,包括冷、热、渗透、盐胁迫、水分缺乏和创伤(图2B),以及与胁迫反应相关的几个其他生物学过程(例如脱落酸信号通路、磷饥饿反应、免疫反应和对卡瑞金的响应)(补充图3)。我们还研究了在 K-Pg 边界期间没有经历古多倍体化事件的其他五个谱系,发现它们保留了 12 个基因家族的小规模重复(补充图4)。然而,这些基因家族主要编码植物代谢过程中的酶或转运蛋白,并不直接与环境适应相关(补充表4)。最近的 WGD 波次发生在 20 Ma 以内,保留了 844 个基因家族的重复(图2A),其功能类别富集在盐胁迫、冷胁迫、水分缺乏和创伤的响应中(图2B)。该时期记录的环境变化包括低 CO2 浓度和相对较凉的气温(Zachos 等,2008)。我们富集的 GO 术语可能部分解释了这些环境变化,但也表明不同谱系可能存在其他环境选择压力。
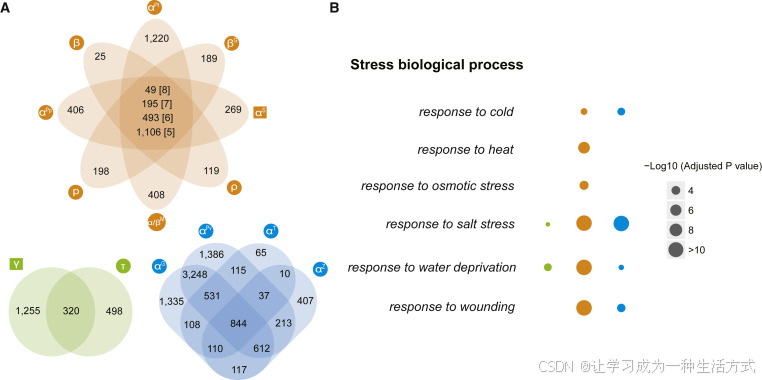
图2. 三个时期中与胁迫相关的基因家族的保留模式
(A) Venn 图显示了在某些时期经历多次 WGD 后,生存下来的共享和特定基因家族的重复情况。数字表示具有基因重复的基因家族数量。方括号中的数字表示经历共享基因家族重复的 WGD 次数。
(B) 显著富集的与胁迫相关的生物学过程的 GO 术语,针对三个时期中共享基因家族保留的基因重复。三个不同颜色的列对应于图 (A) 中的三个时期的 WGDs。
WGD 后转录因子基因家族的偏向保留
转录因子(TFs)在调控许多生物过程(如生长、发育和应激反应)中的基因转录中发挥着至关重要的作用(de Mendoza 等,2013)。以往的研究表明,转录因子是 WGD 后极度保留的基因(Maere 等,2005;Freeling,2009)。
我们根据保留值(R 值,见方法)检查了转录因子基因在三波 WGD 后的重复保留模式。总体而言,大多数转录因子基因在 WGD 后趋向保留(图3),这一点与先前的分析结果一致(Maere 等,2005;Freeling,2009)。然而,我们发现并非所有的转录因子基因家族都被过度保留,而且不同的转录因子家族显示出一定的保留偏好(图3)。例如,具有高保留性的基因家族,包括 ARF、C2H2、C3H、CO-like、ERF、G2-like、GRAS、HD-ZIP、HSF、LBD、MYB、NAC、Trihelix、WRKY、bHLH 和 bZIP 基因家族,通常在不同的进化时期和谱系中反复保留重复基因(图3)。许多高保留的转录因子基因家族参与了多种发育过程以及对非生物和生物胁迫的响应(Khan 等,2018)。然而,有些转录因子在多次 WGD 后的保留较低,如 FAR1、HB-PHD、HRT-like、LFY、LSD、NF-X1、S1Fa-like、STAT、SAP 和 Whirly,这表明这些转录因子的功能和剂量较为保守(图3)。大多数低保留的转录因子基因家族在保守的生物学过程中发挥作用。例如,LFY 控制从营养生长到生殖发育的转换(William 等,2004),而 LSD1 负向调控植物细胞死亡通路(Dietrich 等,1997)。
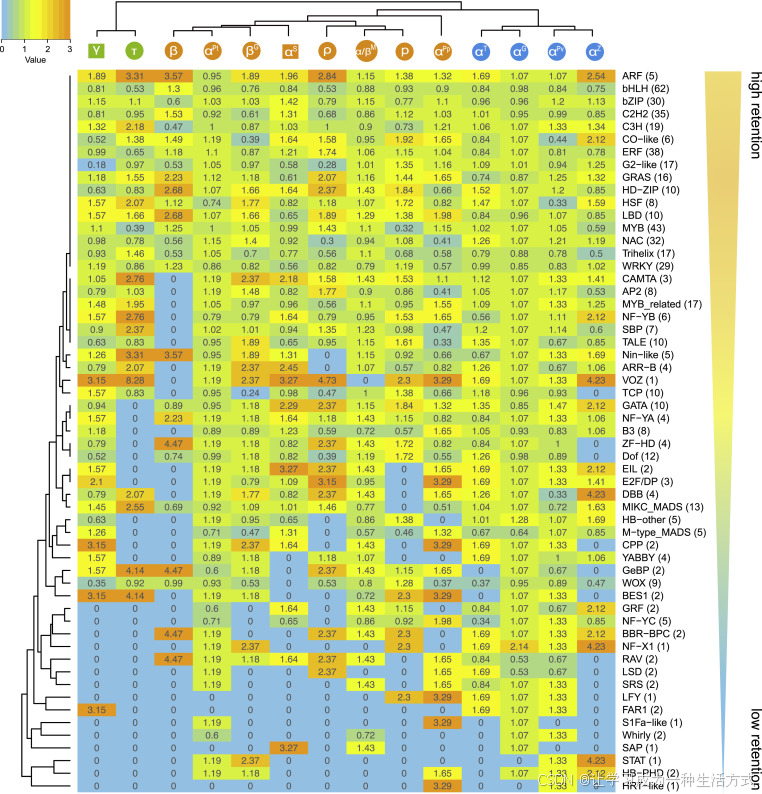
图3. WGDs 后转录因子基因家族的偏向保留模式
转录因子(TFs,行)根据其保留值进行了聚类,WGDs(列)根据其发生时间进行了分组。热图上方的基因家族是经过 WGDs 后保留较高的基因家族,而热图底部的转录因子则是保留较低的基因家族。左上角的颜色键表示转录因子的保留值。热图中每个单元格中的数字表示每个转录因子在相应 WGD 后的保留值。转录因子名称后面的括号中的数字表示属于该转录因子基因家族的同源基因组总数。
那些在特定 WGD 波次后共同保留的重复转录因子基因,被认为是物种在环境变化中生存的关键基因贡献。大约在 120 Ma 时共同保留的转录因子基因主要涉及植物的生长、发育、形态发生以及应激反应(补充表5)。例如,MADS-box 基因家族中四个同源基因组的保留重复基因及其功能分化,可能促进了核心真双子叶植物和单子叶植物花器官的形态新颖性(Zhao 等,2017)。两个热应激转录因子(HSF)同源基因组在应对热应激中起作用。然而,在 K-Pg 边界时共同保留的转录因子主要涉及响应各种非生物胁迫(补充表6)。C2H2、ERF 和 RAV 基因家族的同源基因组参与了对低温的响应。HD-ZIP 家族的同源基因组分别参与了遮阴回避综合症和脱水应激反应。WRKY 家族的同源基因组参与了对低磷应激的响应(补充表6)。
WGDs 对 GRNs 复杂性的贡献
由于 WGDs 可能会重新连接 GRNs(Conant,2010;De Smet 和 Van de Peer,2012),我们旨在探讨 WGDs 对在 K-Pg 边界后适应环境变化过程中网络重塑的贡献(Alvarez 等,1980;Nichols 和 Johnson,2008;Schulte 等,2010)。
全球变冷(或低温)是大规模灭绝时期的主要环境胁迫(Schulte 等,2010),而 C-repeat/DREB 结合因子(CBF)依赖信号通路是公认的主要冷应激信号通路(Chinnusamy 等,2007;Shi 等,2015;Shi 等,2018)。目前,CBF 依赖信号通路的核心组成部分已在拟南芥中解析(Shi 等,2015)。CBF 基因作为通路中的关键组成部分,受上游 ICE 和 CAMTA 转录因子的调控(Shi 等,2015;Zhao 等,2015),并能够在冷胁迫下触发许多冷响应(COR)基因的表达(Chinnusamy 等,2007)。
通过追踪 CBF 通路中关键基因家族的进化历史,我们发现 CBF、ICE、CAMTA 以及其他相关基因家族(如 SIZ、EIN 等)在许多不同谱系中以重复状态出现(图4A)。ICE1 和 ICE2 是从 β WGD 在拟南芥中重复产生的(图4B 和 4C)。ice1 功能缺失突变体对冷胁迫敏感,导致存活率显著低于野生型(Chinnusamy 等,2003)。过表达 ICE2 大大增强了转基因植物的耐冷性(Fursova 等,2009)。在水稻中,CBF 基因在 ρ WGD 后也以重复形式保留,这些基因在冷胁迫中也起着重要作用(补充图5)。因此,来自不同谱系、发生于 K-Pg 边界期间的 WGDs 所保留的重复基因,在很大程度上促成了当前 CBF 依赖信号网络在冷应激耐受性中的拷贝数(可能首先是剂量)和复杂性,这种网络在真双子叶植物和单子叶植物中起着作用(图4B)。
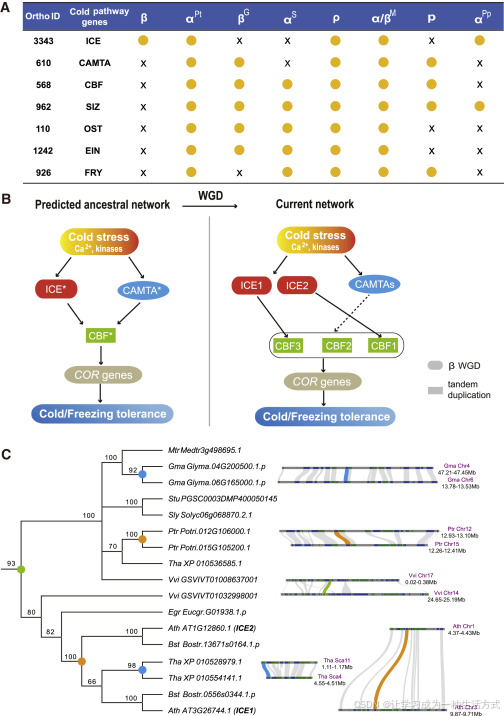
图4. K-Pg 边界周围 WGDs 后冷应答通路关键基因的重复模式
(A) 在 K-Pg 边界发生的八次 WGD 后,已知重要基因家族在 CBF 依赖信号通路中的重复保留状态总结。ICE、CAMTA 和 CBF 是关键的转录因子基因家族,SIZ、OST、EIN 和 FRY 是参与 CBF 依赖信号通路的其他相关基因家族。“x”表示未保留,实心圆点表示基因保留。
(B) 说明了在拟南芥中,WGD 后 CBF 依赖信号通路的扩展和重塑。ICE1 和 ICE2 是由 β WGD 重复产生的。CBF1、CBF2 和 CBF3 由串联重复生成。
(C) ICE 基因家族的系统发育图展示了其进化历史中的基因重复。实心圆圈表示发生在不同阶段的重复。分支上的数字显示了自助法支持值。ICE 基因的同源块位于系统发育树的右侧。
我们还进行了某些谱系的多倍体化事件后 CBF 通路成员网络的比较。共表达网络已广泛用于识别功能相关基因(Obayashi 和 Kinoshita, 2010; You 等, 2016; Obayashi 等, 2018)。为了研究拟南芥谱系中的网络进化,Vitis 是一个理想的外群,因为它在 γ 事件后没有经历额外的 WGD。我们使用分别来自拟南芥和葡萄的 162 和 60 个 RNA-seq 数据,构建了冷应答特异的共表达网络(见方法)。对于来自 β WGD 的重复 ICE 基因,我们检查了拟南芥中的 AthICE1 和 AthICE2 以及葡萄中的同源基因 VviICE 的共表达网络(图5)。大多数 VviICE 模块中的共表达基因与 AthICE1 和 AthICE2 模块中的同源基因聚集在一起,且拟南芥中的相应同源基因可以分为三组:一组特异性与 AthICE1 共表达,一组特异性与 AthICE2 共表达,另一组与 AthICE1 和 AthICE2 都共表达(图5),这表明 WGD 后重复的 ICE 基因发生了亚功能化。此外,拟南芥中 AthICE1 和 AthICE2 模块的规模是 VviICE 模块的两倍,通过在 β 和 α WGD 后将额外的基因招募到网络中,这可能会增强冷应激耐受性。
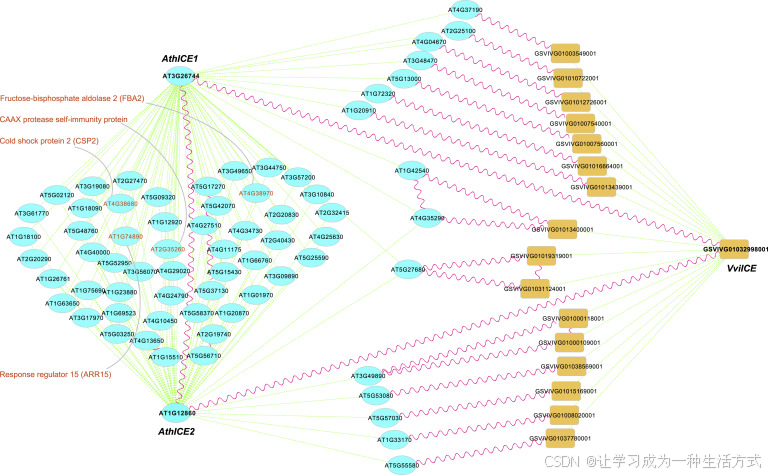
图5. 拟南芥中 ICE1 和 ICE2 重复基因与葡萄中 ICE 同源基因的共表达网络比较
ICE1 和 ICE2 是由β WGD 产生的。红色正弦波线连接相应的同源基因对,它们聚集在同一正交基因组中。两个节点之间的绿色虚线表示正向共表达关系。阿拉伯芥共表达网络中的四个基因,已被证明在冷处理响应中起作用,已突出显示并附有注释信息。
黑暗(或低光)是物种在大规模灭绝时期遇到的另一个主要环境胁迫,这是由于大气尘埃反射阳光,导致长时间的光照不足(Schulte 等, 2010)。我们研究了植物中遮阴回避通路的关键组成部分(Jiao 等, 2007; Ruberti 等, 2012),并发现多个谱系中有几个关键基因是通过 WGDs 重复的(图6A)。在拟南芥中,HD-ZIP II 基因家族中的 ATHB2 和 HAT1,参与遮阴回避反应,源自 β WGD(图6B 和 6C)。分子遗传分析表明,ATHB2 在拟南芥中对低红:远红光的诱导反应非常迅速,athb2 功能缺失突变体显示出较野生型显著减少的胚轴伸长和遮阴回避能力(Carabelli 等, 2013)。以拟南芥为例,图6B 展示了从 WGD 前到 WGD 后网络进化的假设模型。尽管预测的祖先网络存在一定的不确定性,我们的结果提供了清晰的证据,表明在 WGD 后遮阴回避通路发生了扩展,这可能增强了光信号的感知能力,并更好地适应低光环境。
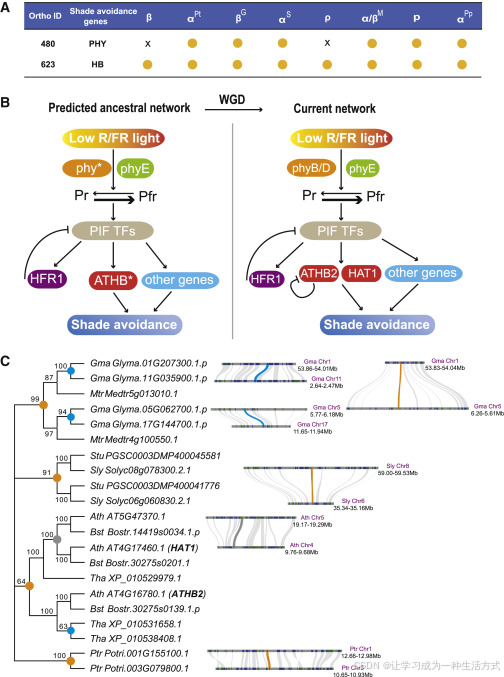
图6. K-Pg 边界周围 WGDs 后遮阴回避通路关键基因的保留模式
(A) K-Pg 边界周围八次独立 WGD 后 PHY 和 HB 基因家族的保留状态总结。PHY 和 HB 是遮阴回避通路中的两个主要基因家族。“x”表示未保留,实心圆点表示基因保留。
(B) 通过将预测的祖先网络与当前 A. thaliana 网络进行比较,说明了 WGD 后遮阴回避通路的扩展与重塑。ATHB2 和 HAT1 由 β WGD 产生。
(C) HD-ZIP II 基因家族的系统发育图,展示了其进化历史中的基因重复。实心圆圈表示不同阶段发生的重复。分支上的数字显示了自助法支持值。HD-ZIP II 基因的同源块位于系统发育树的右侧。
为了检验 WGDs 与植物适应性之间的可能联系,我们比较了在三次 WGD 后,响应冷和黑暗胁迫的调控基因的特定保留模式。冷应激通路的调控基因在最近两次 WGD 后(约66和<20百万年前)具有较高的保留概率,这与这两个时期全球变冷的记录相对应(Nichols 和 Johnson, 2008; Zachos 等, 2008; Schulte 等, 2010)(补充图6)。全球黑暗仅在 K-Pg 边界期间被报道(Nichols 和 Johnson, 2008; Schulte 等, 2010)。与其他两次 WGD 相比,K-Pg 边界周围的 WGDs 后,遮阴回避通路中的基因保留特别高(补充图6)。此外,我们进一步研究了另一个胁迫通路(Na+ 耐受性),尽管高 Na+ 不是 K-Pg 边界期间的主要全球胁迫。盐过敏(SOS)信号通路在维持 Na+ 高耐受性下的离子稳态中发挥作用(Ji 等, 2013;补充图7A)。SOS 通路核心成员的重复基因,如 SOS3、ScaBP8、SOS2 和 SOS1,仅在所检查的 WGDs 后偏向性地保留(补充图6和7B)。因此,多次独立 WGD 后应激相关网络中关键成员的优先保留,可能作为支持 WGDs 促进物种在全球环境变化中适应的重要证据。
讨论
古代 WGDs 在被子植物中的周期性发生特征
为了弥补古代多倍体化对适应性进化的遗传贡献之间的差距,我们需要探索在被子植物进化历史中多次 WGD 的实证适应性遗传特征。多倍体在自然界中非常常见。然而,初期多倍体个体往往面临内部和外部障碍,包括染色体分离错误率增加、有效种群大小小、与亲本二倍体物种的竞争等(Comai, 2005; Arrigo 和 Barker, 2012)。几项研究表明,多倍体通常是进化的死胡同(Stebbins, 1950; Mayrose 等, 2011)。新形成的多倍体植物必须找到不同于二倍体物种的生态位才能生存(Stebbins, 1950; Levin, 1983; Ramsey, 2011; te Beest 等, 2012; Visger 等, 2016)。多倍体植物可能会迁移到一个新的、充满压力的环境中,在那里没有与祖先二倍体的竞争,或者它们可能在强烈的环境选择后生存下来,将二倍体祖先替换为多倍体(Otto 和 Whitton, 2000; Brochmann 等, 2004; Ramsey, 2011; te Beest 等, 2012; Chao 等, 2013; Parisod 和 Broennimann, 2016)。因此,被子植物中的古代多倍体化事件似乎与过去的全球环境变化聚集并共同发生,这可能在多倍体的建立中起到了重要作用(Van de Peer 等, 2009; Van de Peer 等, 2017)。
推断 WGDs 在进化意义上的挑战
由于 WGDs 反复发生并伴随大规模基因丢失,古代 WGDs 的遗传贡献信号变得复杂且模糊(Doyle 等, 2008; Schnable 等, 2011; Wendel 等, 2016)。此外,环境选择压力通常不会持续数千万年。古代 WGDs 的新遗传贡献可能在环境条件变化后丧失。此外,杂交和重组也可能会去除帮助物种在特定时期生存下来的关键信息。因此,推断 WGDs 在被子植物进化历史中的意义是充满挑战的。为了解决这个问题,我们在分析中包括了许多高质量的完全测序基因组,它们共享一个古代 WGD,以避免由于基因组组装和注释不完整或不当而错过关键基因。更重要的是,尽管某些物种可能丧失了这些关键基因,但我们仍然可以通过同时考虑多个物种来拼凑出一个广泛的图景。最后,我们研究了在同一时期发生的几个独立 WGD,以寻找共享的重复基因。通过这种方法,我们能够识别出支持物种在剧烈环境变化中生存的关键遗传信号,并提出 WGDs 对植物适应性进化的可能影响。
遗传证据为 WGDs 促进适应性贡献提供新视角
为了说明单个 WGD 的遗传影响,我们还需要独立考虑在不同谱系中发生的某些古代 WGD 波。剧烈的环境变化应为地球上所有物种施加了相似的选择压力。先前的研究表明,涉及调控和发育的基因存在偏向性保留(Maere 等, 2005; Freeling, 2009)。通过全面研究基因家族,我们发现,在相同时期发生的独立 WGD 后,某些功能基因发生了重复,这为全球环境选择对不同谱系的古代多倍体提供了支持的证据。例如,响应 K-Pg 边界期间低温和低光环境变化,第二波古代 WGD 有助于重塑 CBF 依赖的信号通路(图4和图5)和遮阴回避通路(图6)。ICE 和 CBF 基因家族的重复被招募到通路中,确实增强了植物的耐寒性(Shi 等, 2015)。
方法
基因组数据
我们选择了25个已测序的植物基因组,代表了被子植物的主要谱系,并且在其进化历史中有明确的WGD记录。研究的物种包括10个真双子叶植物(A. thaliana、Boechera stricta、Eucalyptus grandis、G. max、Medicago truncatula、Populus trichocarpa、Solanum lycopersicum、Solanum tuberosum、Tarenaya hassleriana和V. vinifera)、12个单子叶植物(Brachypodium distachyon、O. sativa、P. virgatum、Sorghum bicolor、Setaria italic、Spirodela polyrhiza、Setaria viridis、Z. mays、Aegilops tauschii、Elaeis guineensis、Hordeum vulgare和M. acuminata)、一个现存代表的基底被子植物(A. trichopoda)、一个蕨类植物(S. moellendorffii)和一个苔藓植物(P. patens)。A. tauschii、E. guineensis 和 M. acuminata 的基因组数据从其项目网站下载(补充表1),其他基因组数据主要从 Phytozome(版本11)下载(Goodstein 等, 2012)。
基因家族分类与系统发育分析
我们使用 OrthoMCL 方法(版本 2.0.9)(Li 等, 2003)将蛋白编码基因分类为假定的基因家族或亚家族,采用膨胀参数 1.5,获得总共 66,509 个正交基因组。少于四个基因和/或没有来自外群基因的基因的正交基因组被过滤掉,剩余的 12,077 个正交基因组被用于系统发育分析。通过考虑每个正交基因组中基因的最后共同祖先,展示了 12,077 个正交基因组的分类分布(补充图 8)。为了构建全基因家族树,每个正交基因组的氨基酸序列使用 MAFFT 对齐(Katoh 等, 2005)。然后,使用 PAL2NAL 将相应的核酸序列强制映射到氨基酸对齐(Suyama 等, 2006)。为了去除对齐不良区域,使用 trimAl 1.4(Capella-Gutirrez 等, 2009)和“automated1”选项精细调整了核酸对齐。系统发育树使用最大似然法在 RAxML 8.2.11 中进行(Stamatakis, 2014),采用快速自助法选项,进行 100 次重复计算,使用 GTRGAMMA 模型。
鉴定基因重复事件
为了准确识别基因重复事件,我们遵循了 Jiao 等(2011)提出的基因树与物种树的和解标准。即,两个子分支需要至少有一个来自共同物种的基因,且父节点和其中一个子节点的自助法支持值均应不小于 50%。由于本研究中采样的25个物种的系统发育关系已明确(Angiosperm Phylogeny Website),我们直接采用当前接受的物种树拓扑。外群物种(P. patens、S. moellendorffii 和 A. trichopoda)的基因用于根化树。
首先,我们使用 Notung 2.9(Stolzer 等, 2012),一种基因树-物种树和解程序,批量和解所有基因树的节点与物种树中对应的节点。Notung 使用基于简约的优化标准,以最小化重复/丢失成本。我们根据重复-丢失事件模型运行分析。除了“—reconcile”模式外,我们还使用“—rearrange”模式,并设置参数为“—threshold 50%”。该选项可以重新排列弱支持的边(如自助法支持值<50%),并减少推断中重复的不确定性(Notung 2.9 手册)。其次,在仔细检查了 Notung 结果的成千上万的和解树后,我们进一步应用了“两个子分支需要至少有一个共同物种的基因”这一标准,并从所有和解树中移除了部分低置信度的重复。
消除串联重复
我们定义位于五个基因以内的两个基因为串联重复。如果一个重复节点包含两个基因(基因1、基因2),或两个子分支((基因1、基因2)、(基因3、基因4)),且这两个基因彼此靠近,则视为串联重复。根据上述标准,我们为所有的和解基因树移除了串联重复事件。
重复节点的 KS 计算与个别 WGDs 的界定
我们使用 KS 值来对无法通过系统发育方法界定的在同一分支上发生的两个 WGD 事件进行定年。通过 PAML 包中的 yn00 程序(Yang, 1997)使用 Nei-Gojobori 方法(Nei 和 Gojobori, 1986)获得成对比较的 KS 估计(一个基因来自 m 分支,另一个基因来自 n 分支)。然后,将所有成对比较的 KS 值之和除以 KS 估计的数量(m∗n),从而得到重复事件的加权 KS 值。根据 O. sativa 的同源基因对的序列 KS 曲线,我们大致定义 KS ≥ 1.0 的重复节点为 σ 事件,KS < 1.0 的重复节点为 ρ 事件(补充图9)。KS ≥ 0.7 属于 γM 事件,KS < 0.7 属于 α/βM 事件,这一界定基于先前的研究(D'Hont 等, 2012)。
保留同源基因的同源性分析
上述程序使我们能够获得与每个 WGD 相关的同源基因。为了进一步验证重复基因是否仍位于同源块中,我们对 WGD 衍生物种进行了共线性分析。通过使用 MCScanX,基于默认参数检测基因组内的同源块(Wang 等, 2012)。然后,我们通过系统基因时间计算了具有同源证据的同源基因在总基因中的百分比。
GO 注释与功能富集分析
为了注释正交基因组,我们使用 A. thaliana 基因的完整 GO 术语作为正交基因组的注释,如果它们具有拟南芥基因。否则,我们使用 InterProScan(Zdobnov 和 Apweiler, 2001)搜索蛋白序列的 InterPro 域并获得完整的 GO 术语注释。通过 Fisher 精确检验,比较样本(共同保留的正交基因组)与背景(所有注释过的正交基因组),并根据 Benjamini 和 Hochbery(假发现率)方法调整 P 值,评估 GO 术语的统计富集。
转录因子的保留分析
我们使用从 PlantTFDB 4.0(Jin 等, 2017)下载的拟南芥转录因子(TF)基因家族来注释正交基因组。对于每个 WGD 事件,识别每个 TF 家族的保留正交基因组。由于重复后序列的分化,一些 TF 家族通常被归类为多个正交基因组。为了消除同一 TF 基因家族中正交基因组大小的影响,我们计算了一个标准化值(保留值,R 值)来反映每个 TF 在相应 WGD 后的保留模式。R 值通过以下公式计算:

共表达网络构建与比较
我们从 NCBI 的 Sequence Read Archive 数据库(https://www.ncbi.nlm.nih.gov/sra/)下载了222个冷应激相关的 RNA-seq 样本(162个来自拟南芥,60个来自V. vinifera)(补充表7和8)。然后,对这些数据集进行质量控制、清洗、FPKM(每百万映射的片段每千碱基的片段数)计算,如同 You 等(2016)所述。在移除每个样本中非表达基因后(拟南芥和V. vinifera的FPKM截止值分别为0.14和0.36;详细方法见You等,2016),剩余的表达基因用于计算共表达关系,使用 Pearson 相关系数(PCC)。随后,使用互信息排序(MR)(计算为基因A到基因B的PCC排序的几何平均值与基因B到基因A的排序),用于构建共表达网络(Obayashi 和 Kinoshita, 2010; You 等, 2016)。MR被证明比PCC更有效地获得可信的共表达基因对(Obayashi 和 Kinoshita, 2010)。因此,我们为每个物种构建了基于MR的共表达网络。然后,我们选择了拟南芥和Vitis中最前面300个共表达基因(You等[2016]和Obayashi等[2018]所使用的阈值),用于网络比较。在这种情况下,拟南芥中由β WGD形成的AthICE1和AthICE2及其在Vitis中的同源基因VviICE用于评估这些关键基因在CBF信号通路中的进化模式。