在线计算“蛋白-蛋白复合物”的MM/GBSA
VD-MM/GBSA 是 HawkDock2 平台集成的结合能预测方法,旨在高精度计算蛋白质-蛋白质复合物的结合自由能,解决传统方法在预测亲和力时的局限性,为复合物相互作用强度评估提供更可靠的量化依据。
HawkDock2平台集成的MM/GBSA以及VD-MM/GBSA方法为了在降低计算成本的同时保证准确性做出了一定的调整:
① MM/GBSA:
• MM项:
基于 ff02 力场描述原子间相互作用,包括键合作用(共价键、角度、二面角)和非键合作用(范德华力、静电作用)。• 广义 Born(GB)模型:
使用 GBOBC1 模型描述溶剂化效应,内部介电常数设为 1(模拟非极性环境),通过计算广义 Born 半径近似溶剂对静电作用的屏蔽效应。• 表面积(SA)项:
考虑溶剂暴露表面积与表面张力的关系,描述疏水相互作用对自由能的贡献。
② VD-MM/GBSA
VD-MM/GBSA 是上述方法的改进版,根据残基类型设定不同介电常数(传统 MM/GBSA 介电常数固定为 1),更精确模拟蛋白质内部极性与非极性环境的差异,提升结合自由能预测精度。
MM/GBSA 计算前仅对系统进行 5000 步能量最小化(2000 步最速下降法 + 3000 步共轭梯度法),目的是消除结构中不合理的原子碰撞或高能量构象,得到能量最低的静态结构。该方法未通过 MD 模拟获取动态构象集合,仅基于单一优化后的构象计算结合自由能及残基贡献。
网站地址
http://cadd.zju.edu.cn/hawkdock/
使用方法
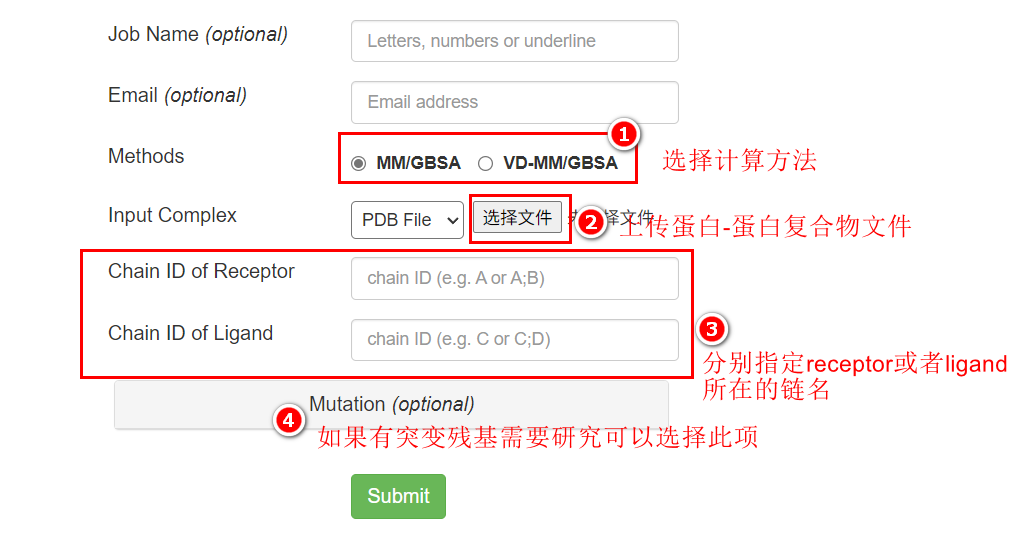
• ① 上传的复合物pdb文件只能是蛋白-蛋白复合物,不要糊里糊涂地上传蛋白-小分子或者蛋白-核酸!!!
• ② 由于分析的是蛋白-蛋白复合物,所以无所谓哪个组分当receptor或者ligand, 可以自己指定。比如复合物两个蛋白的链名分别是C, E,那么我既可以选择C链当receptor,也可以选择C链当ligand。
• ③ 如果想评估突变残基对结合能的影响,可以选择上图的步骤4,点击后会弹出如下页面:

根据自己研究的实际情况输入receptor或者ligand组分上的突变信息,突变信息的输入应遵循如下语法:
链ID-残基ID-突变前残基名-突变后残基名
举个例子:
B-16-ARG-MET表示B链的16位精氨酸(ARG)残基突变为甲硫氨酸(MET)
结果展示
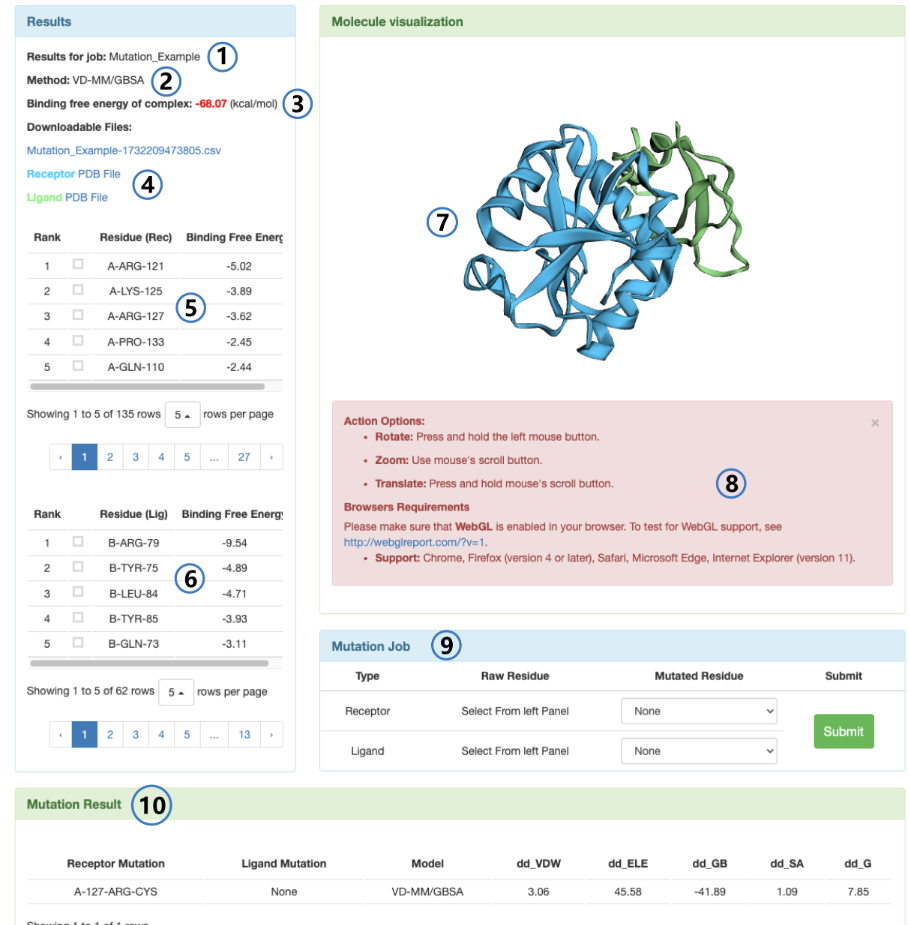
• ① 任务名称:显示当前任务的名称。
• ② 计算方法:标注本次计算使用的方法(MM/GBSA 或 VD-MM/GBSA)。
• ③ 复合物结合自由能:蛋白质 - 蛋白质复合物的结合自由能数值。
• ④ 可下载文件
• ⑤ 能量贡献表格
按能量贡献从大到小排列的受体残基列表,支持用户选择残基在右图中可视化。• ⑥ 配体残基的能量贡献表,格式与受体表一致。
• ⑦ 分子可视化
基于 3Dmol.js 的可视化窗口,展示复合物三维结构。• ⑧ 附带操作说明(鼠标交互方式及浏览器要求)。
• ⑨ 突变任务面板
支持选择受体 / 配体的野生型与突变残基,提交突变任务。
实时显示任务状态与结果。• ⑩ 突变结果面板
展示各突变任务中残基突变引起的能量变化。
参考文献
Zhang X, Jiang L, Weng G, et al. HawkDock version 2: an updated web server to predict and analyze the structures of protein-protein complexes. Nucleic Acids Res. Published online May 6, 2025. doi:10.1093/nar/gkaf379