文献阅读 | Briefings in Bioinformatics | Hiplot:全面且易于使用的生物医学可视化分析平台
文献介绍

文献题目: Hiplot:一个综合且易于使用的 Web 服务,用于增强出版物准备的生物医学数据可视化
研究团队: Openbiox/Hiplot 社区
发表时间: 2022-07-05
发表期刊: Briefings in Bioinformatics
影响因子: 13.99(2022年)
DOI: 10.1093/bib/bbac261
摘要
在临床(clinical)、组学(omics)和机制(mechanism)实验过程中产生的复杂生物医学数据,正日益通过基于云计算和可视化的数据挖掘技术得到深入开发。然而,科学界目前仍缺乏一个易于使用的网络服务平台,用于生物医学数据的全面可视化,尤其是能够根据用户需求灵活缩放、随时更新且达到期刊出版级质量的可视化图表。为此,作者推出一个社区驱动的现代化网络服务,Hiplot(https://hiplot.org),为生命科学和生物医学领域提供简洁优质的数据可视化应用。该平台使用户能便捷地通过交互方式完成若干专业化可视化任务,而这些任务以往只能由资深生物信息学或生物统计学研究者完成。该服务配备 240+ 生物医学数据可视化功能,涵盖基础统计、多组学分析、回归分析、聚类分析、降维分析、荟萃分析、生存分析、风险模型构建等方向,可满足生物医学研究者的大部分日常需求。为提升插件使用和开发效率,作者在网站客户端/服务端引入了多项核心优势:基于电子表格的数据导入、跨平台命令行控制器(Hctl)、多用户管道计算集群、基于 JSON 的插件系统、简易的数据/参数-结果-错误复现机制以及实时更新模式。通过演示数据集/真实数据集和基准测试,作者基于精选原生插件对统计参数、癌症基因组图谱、疾病风险因素及网站性能进行了系统评估。平台访问量和用户数量的统计数据进一步印证了该服务对相关领域的潜在影响力。这项新兴的免费网络服务将为生命科学与数据科学领域的研究者提供重要支持。
前言
对源自实验检测、组学研究和临床观察的多维生物医学数据进行探索与挖掘,主要依赖于现代图形学与统计学方法,例如疾病诊断和统计描述/推断。提升假设驱动型与数据驱动型科学研究的可解释性、可重复性和有效性,正是可视化数据挖掘技术应当发挥的关键作用。十余年前,用户只能通过功能有限、扩展性不足的桌面应用程序进行日常科研数据可视化与分析。而如今,具备更优扩展性的基于云的网络应用,已成为缺乏编程技能的生物学家和临床工作者处理复杂生物医学数据的理想选择。并且,随着 Galaxy、DNAnexus 等知名生物信息学云服务的建立,序列比对、变异检测、表观遗传分析等基于流程的上游数据分析任务已得到一定简化。
然而在这些平台中,基于表格数据的出版级科学图表与交互式数据挖掘等典型下游功能仍显不足。例如知名生物信息云平台 Galaxy 虽提供数十个可视化插件,但对轻量级生物医学可视化任务的优化仍不充分;St. Jude 儿童研究医院云门户的可视化模块虽有 20 个基于 JavaScript 的癌症基因组交互插件,却缺乏基础科研图表功能;而 imageGP 自 2017 年以来仅开发了 16 项科学图表与分析子功能。要满足多样化的可视化需求,仍需科学界通力合作。研究显示,复杂的用户界面和薄弱的交互性已成为阻碍用户采用生物医学数据可视化工具的主要因素。例如,现有网络工具鲜少支持在线电子表格的数据预览与编辑功能,对出版级排版中多图自动排列(如每页4/6/9图)也普遍欠缺考量。此外,任务输出延迟、缺乏跨平台命令行工具、数据/参数-结果-错误复现困难等问题,进一步限制了网络工具在生物医学可视化中的广泛应用。
值得注意的是,在整体网站设计中需通过核心功能优化网络服务,以提升效率并降低时间成本。同时,构建全面可扩展的可视化网络服务面临的关键挑战在于:如何简化用户操作与开发者工作流程。这需要精心设计的前端客户端/命令行工具、高质量输出生成、高效完成基础任务的能力、完善的用户支持服务以及低门槛的数据/参数-结果-错误复现机制。此外,平台还需通过长期维护持续更新,提升插件的可用性与定制化程度。建立一个专注于出版级生物医学可视化的开放协作社区,将加速复杂生物医学网络应用的构建、验证与更新。
为应对这些挑战,作者推出了易于使用和可扩展的网络服务,Hiplot(https://hiplot.org)与跨学科社区Openbiox(https://openbiox.org),专注于生物医学数据可视化交互应用的开发。自 2019 年 10 月以来,Hiplot/Openbiox 协作组已开发数百个基于可视化的数据挖掘交互插件。Hiplot 核心功能基于开源方法构建,覆盖生物学家与临床工作者最常见的日常可视化需求,并采用简洁 UIs 与高效交互设计。例如可切换的编辑表格与文件上传/路径选择视图,显著提升了数据导入、预览与导出效率。同时,基于多用户管道计算集群的响应策略结合版本控制,大幅降低了基础科研图表生成的时间成本。而且,平台还实现了基于 JSON 的结构化插件系统,包含预置 UI 组件、R 开发库和在线预览器,以持续扩展功能。通过原生插件的基准测试及访问量/用户数统计,作者评估了平台性能,这些数据将为 Hiplot 及其他同类网 web/R 工具的迭代更新提供重要参考。作者期待这个实时更新的工具包能发展成为生物医学、生命科学与数据科学领域重要的可视化基础设施。
研究结果
1. Hiplot 的全面功能和优势概述
自 2019 年以来,Hiplot/Openbiox 联盟已开发了大量基于网络的交互式可视化应用(240+),用于生物医学数据挖掘(Figure 1, Table S1)。另外,通过系统的文献调研,本研究被证实是为数不多通过社区协作建立的免费网络服务之一,能够交互式、全方位地生成出版级生物医学可视化图表(Table 1)。观察显示,Hiplot 平台(native Hiplot、R Shiny、Python Streamlit)提供的交互式可视化工具在数量与多样性上均优于同类平台。例如,该网络服务可提供与知名商业科学绘图软件 GraphPad 相媲美的现代统计图表功能(Figure 1)。因此,用户无需受操作系统或软件环境限制,即可通过这些开放获取的可视化功能处理日常数据分析需求,包括数据相关性、分布特征、百分比构成、演化趋势、流动关系、排序比较及空间特征等维度的展示与统计推断。

Figure 1. Hiplot web 服务中核心功能和优势的概述
顶部面板展示了网站核心功能分类及其相关受益领域。该平台已构建了涵盖现代统计图表、组学分析和临床模型的完整生物医学可视化功能体系,用户可直接使用 Hiplot 生成符合出版要求的高质量可视化图表。图库展示了基础图表、组学数据和临床模型相关插件的部分应用案例及输出效果,更多示例可直接在网站应用卡片列表中查看。底部面板突出了提升使用/开发效率和用户体验的核心优势:基于电子表格和可切换文件上传器的设计简化了网络轻量化可视化任务的数据导入流程;依托多用户管道计算集群可在数秒内完成基础图表生成,同时支持分布式部署和 R 运行环境的版本控制;采用基于 JSON 的插件系统及预置UI/后端功能模板,有效减少了轻量化生物医学可视化任务的冗余开发步骤;网站根据用户建议实时更新,已解决的个性化需求帮助设置了更多控制图形输出的自定义参数。
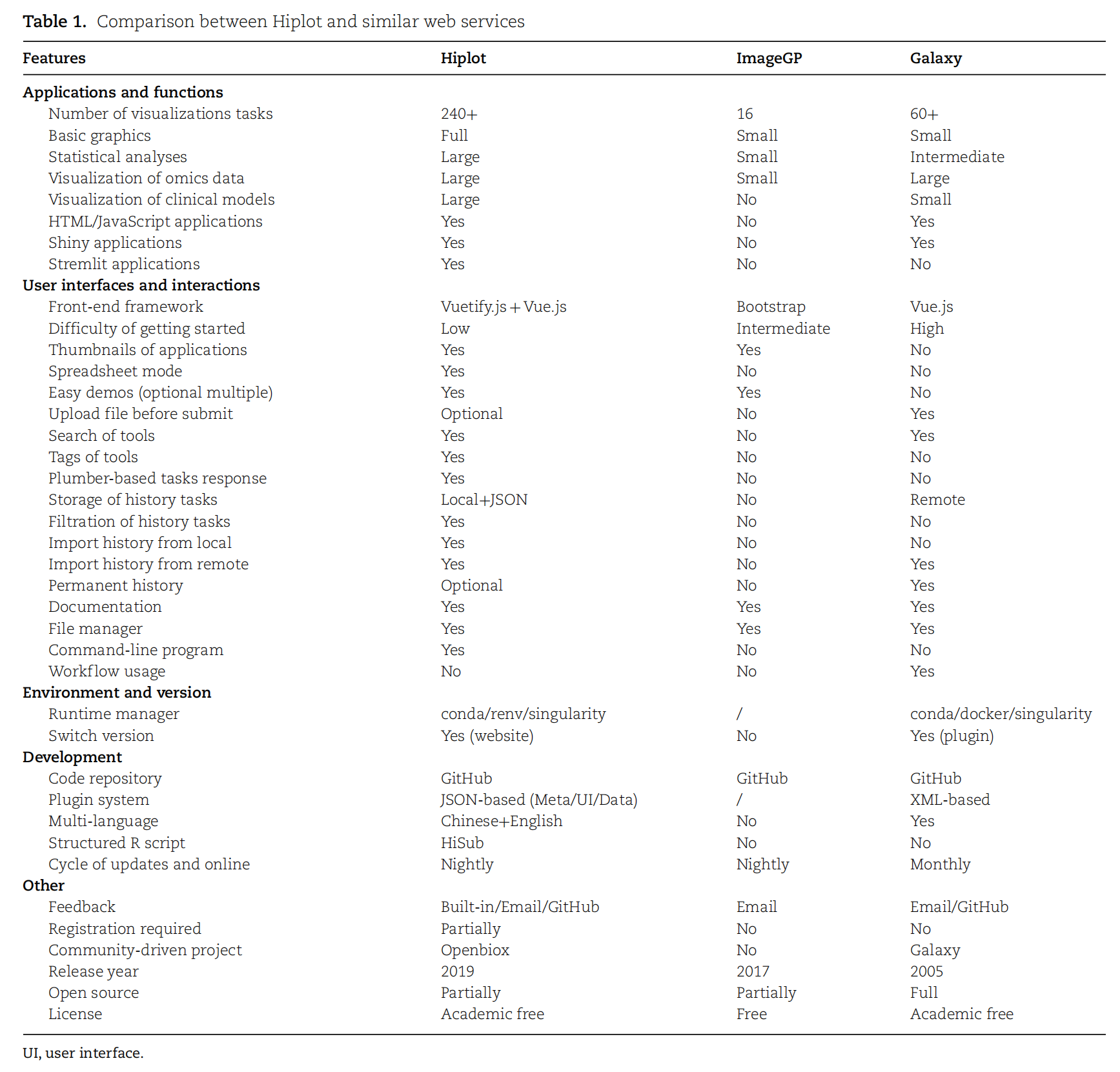
Table 1. Hiplot 与类似 web 服务之间的比较
在 Hiplot 中实现的诸多基础可视化改进已吸引了科学界的广泛用户,同时平台还提供组学与临床数据可视化功能,为生物医学和生物学研究者提供更深入的支持。如今,用户可在 Hiplot 网站上自由交互式探索癌症多组学数据集,全面实现基因组结构、染色体分布、遗传变异、群体遗传学、基因表达谱、基因通路富集和肿瘤微环境(TME)等多组学数据可视化(Figure 1)。此外,平台还提供基于机器学习的可视化方法,包括无监督聚类、降维算法(DRAs)、线性/非线性回归、荟萃分析、生存分析和风险模型等,使用户能够关联多维特征并开展转化研究(Figure 1)。
除了全面的功能(基础科学图表、组学数据可视化、临床模型)外,Hiplot 的其他主要优势还包括:简洁的设计与交互方式(如基于电子表格的数据表)、丰富的输出样式与配色主题、基于 Plumber 的多用户后端任务框架、基于 JSON 的插件系统、便捷的任务复现机制以及功能的实时更新(Figure 1)。Hiplot 采用的现代化用户界面和高效交互方式显著降低了用户学习成本。用户可通过主网页客户端或命令行程序调用 Hiplot 的丰富功能(Figure 2A)。平台通过标签搜索的关键词模糊匹配功能,可快速定位基础工具、高级工具、临床工具和迷你工具等不同模块的网页插件,顶部路径栏也支持插件切换跳转。此外,卡片缩略图能帮助用户快速识别所需工具。相较于传统生物信息学网站仅支持文件路径选择或文本区域上传的模式,Hiplot 插件中具有更佳可读性和可编辑性的电子表格数据编辑器,可能成为同类网站的可选替代方案。同时,海量插件提供的丰富配色、主题和输出选项,使用户能轻松生成个性化结果。基于 Plumber 的多用户响应机制相比需要重新加载依赖项的工作流式响应方法,进一步提升了轻量化可视化任务的执行效率(Figure 2B)。来自本地或远程存储的标准 JSON 数据对象可在数秒内复现插件的输入输出。本研究实现的基于 JSON 的插件系统,使作者能够开发这个涵盖多研究领域的大规模网站——特别是结构化的 UI 组件描述通过提供多插件共享的字段值控制组件/后端功能,简化了冗余开发工作。网站提供的 JSON 文件还支持用户在浏览器和命令行环境中轻松复现任务。此外,网站功能的实时更新将加速 Hiplot 的开发周期。虽然目前尚未支持工作流编辑器设置任务,但通过命令行程序将 Hiplot 功能整合至现有数据分析流程是可行的。若未来用户需求强烈,可利用现有插件构建类 Galaxy 界面以实现更优的工作流整合。
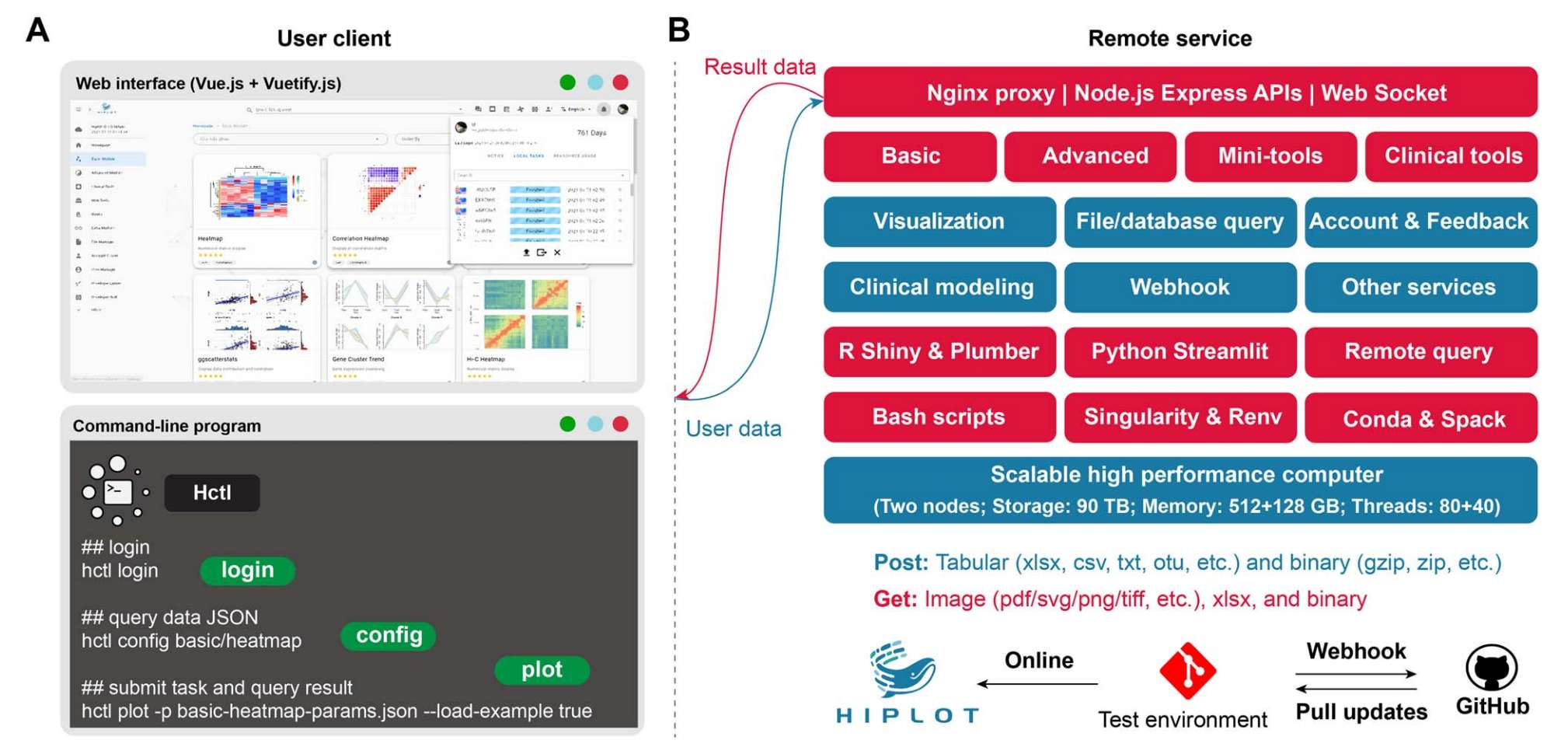
Figure 2. 网站基础架构和从客户到后端服务的组件
(A) Hiplot 的网页客户端与命令行程序 Hctl。顶部窗口展示的是 Hiplot 网页客户端在基础模块页面的截图,其中列出了若干基础应用卡片。页面左侧设有主导航菜单,方便用户在不同模块间切换浏览,同时通知窗口会显示历史任务记录。底部窗口则展示了 Hctl 的子命令功能,包括登录(login)、配置(config)和提交(submit)。使用 Hctl 程序需先完成登录,config 子命令用于查询演示数据/参数,submit 子命令则用于提交任务。
(B) 基础设施示意图呈现了 Hiplot 网络服务的核心后端服务与硬件资源架构。Hiplot 的网页端和命令行客户端通过 Nginx proxy / Node.js Express API / Web Socket services 进行通信。平台的任务插件分布在四大核心模块:基础工具、高级工具、迷你工具和临床工具。除基于 JSON-based Vue.js插件外,还引入了 R Shiny 和 Python Streamlit 框架来构建交互式应用。Hiplot 的运行环境由 conda、spack、Singularity、renv 进行管控,当 Github 上的 Hiplot 或 Openbiox 代码库有新提交时,相关插件会自动同步至开发或生产环境进行验证。
2. 使用案例
由于篇幅所限,作者仅基于示例/真实数据集介绍少量具有代表性的功能插件。这将有助于读者理解网站的功能分类和基本使用方法。这些用例主要分为三大部分:基础科学图表、癌症组学数据可视化以及临床模型。
使用案例1:基础科学图表
作者选取 Hiplot 网站上使用频率最高的科学图表——热图(Heatmap),来展示基础可视化任务的常规操作流程(Figure 3A)。用户进入插件页面后,可通过点击顶部/底部的演示按钮加载示例数据,查看标准输入格式。需特别注意的是,热图插件要求输入数值型数据表(行代表特征,列代表样本)及可选的行/列注释信息。这些数据表支持通过剪贴板粘贴、本地文件上传或远程文件服务器三种方式导入。对于超过 2MB 的基因表达矩阵文件,建议使用文件上传器选择路径模式进行传输。热图插件提供通用参数用于控制图表宽度、高度、字体、主题、行列注释的配色方案以及行列文本和标题的字体大小。额外参数还包括热图配色、数据标准化方式、高变异特征筛选、聚类方法、距离度量及数值显示等设置。当前默认采用欧氏距离(Euclidean distance)和 ward.D2 算法进行聚类分析,用户可尝试不同聚类方法与距离度量组合以获得最佳效果。当输入特征数量庞大时,选择高变异特征有助于进行子集的无监督层次聚类。若颜色梯度区分度不足,用户可通过"scale"参数选择按行或列进行数据标准化。任务提交后,数据流会经过 Base64 编码传输至后端服务(Figure 3B),由可用的 Plumber 工作节点进行处理。最终生成的热图结果(如 JPG/PDF 格式)将在底部预览窗口显示并提供下载。用户可将参数导出为本地 JSON 文件以便后续快速加载,登录用户还可通过预览窗口的同步按钮将结果永久保存至云端文件管理器。
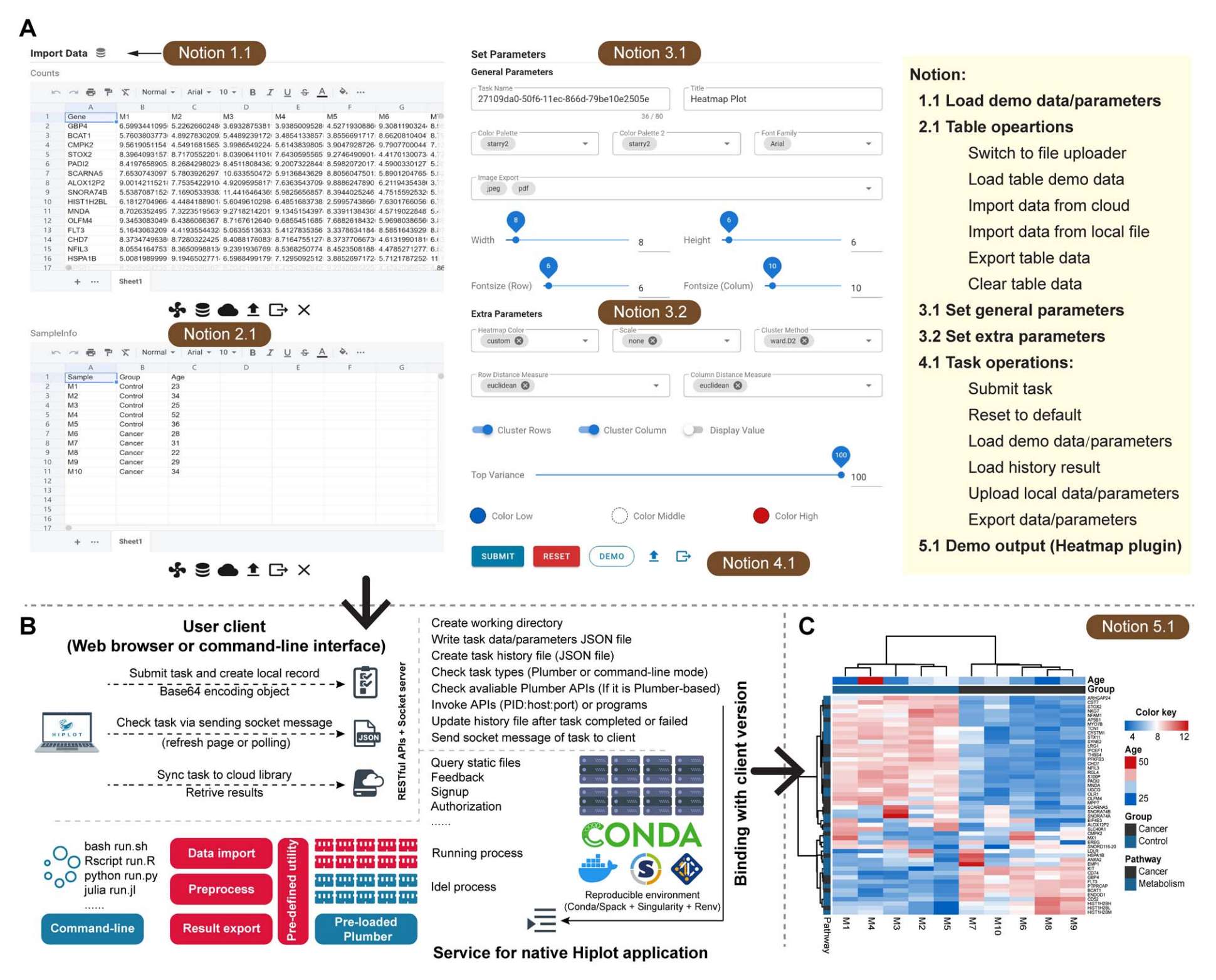
Figure 3. 热图插件的使用流程和任务处理步骤
(A) 热图插件的网页界面及提交新任务的主要操作步骤。表格数据可通过电子表格网页组件进行导入、预览和编辑。右侧面板列出了热图的通用参数和高级参数,这些参数可控制数据预处理、聚类步骤及热图输出样式。其中 "top variance" 参数可用于筛选部分基因进行聚类分析。所有参数均以 JSON 格式存储,可直接导出用于复现输入设置。用户还可通过云端文件管理器的历史记录功能重现输入输出数据。
(B) 后端处理任务的服务流程示意图。提交的数据会经过 Base64 编码,随后由可用的 Plumber 工作节点执行热图插件的核心代码。任务完成后,后端将返回包含任务历史信息的 JSON 文件,以及用于获取图像和记录输出的路径。
(C) 热图插件的示例输出效果。每列代表一个样本,每行代表一个基因。示例输入数据可清晰区分两个聚类(癌症组与对照组)。行列注释分别显示了基因分类(癌症相关或代谢相关)和样本类型(癌症或对照)。
网站基础模块提供了其他基于表格数据的基础科学图表,可用于探索相关性、数据比例或拓扑结构、分布特征、演化趋势、网络关系和空间特性等,这些图表均基于 Hiplot 原生框架构建。此外,迷你工具模块还提供实用的细分功能,可简化数据表中分类变量与连续变量的统计分析,并支持将 Hiplot 生成的图表或本地 PDF 文件进行多图组合。
使用案例2:基于组学的癌症数据可视化
Hiplot 平台已开发了数十个多组学数据可视化插件,尤其专注于癌症基因组学和转录组学分析,主要包含 Shiny 应用程序和原生 Hiplot 插件。2019 年,Openbiox 社区启动了开源项目 UCSCXenaShiny(Figure 4A),该项目通过 R 函数和基于 Shiny 的网页界面,使用户能够搜索、下载、探索并快速分析/可视化来自 UCSC Xena 数据中心的基因组数据集。作者采用 UCSCXenaShiny 的泛癌模块分析表明,促甲状腺激素释放激素的高表达与癌症基因组图谱(TCGA)数据库中急性髓系白血病患者(预后改善)和胶质瘤患者(预后较差)均存在显著关联(Figure S10)。该交互式应用还能探索其他任何基因表达异常或存在序列突变的基因在主要癌症类型中的预后意义。用户可通过点击 Hiplot 网站高级模块中的"Shiny"及相关标签,发现更多基于 Shiny 的癌症组学可视化应用,例如全基因组关联研究相关的 Shiny 插件。
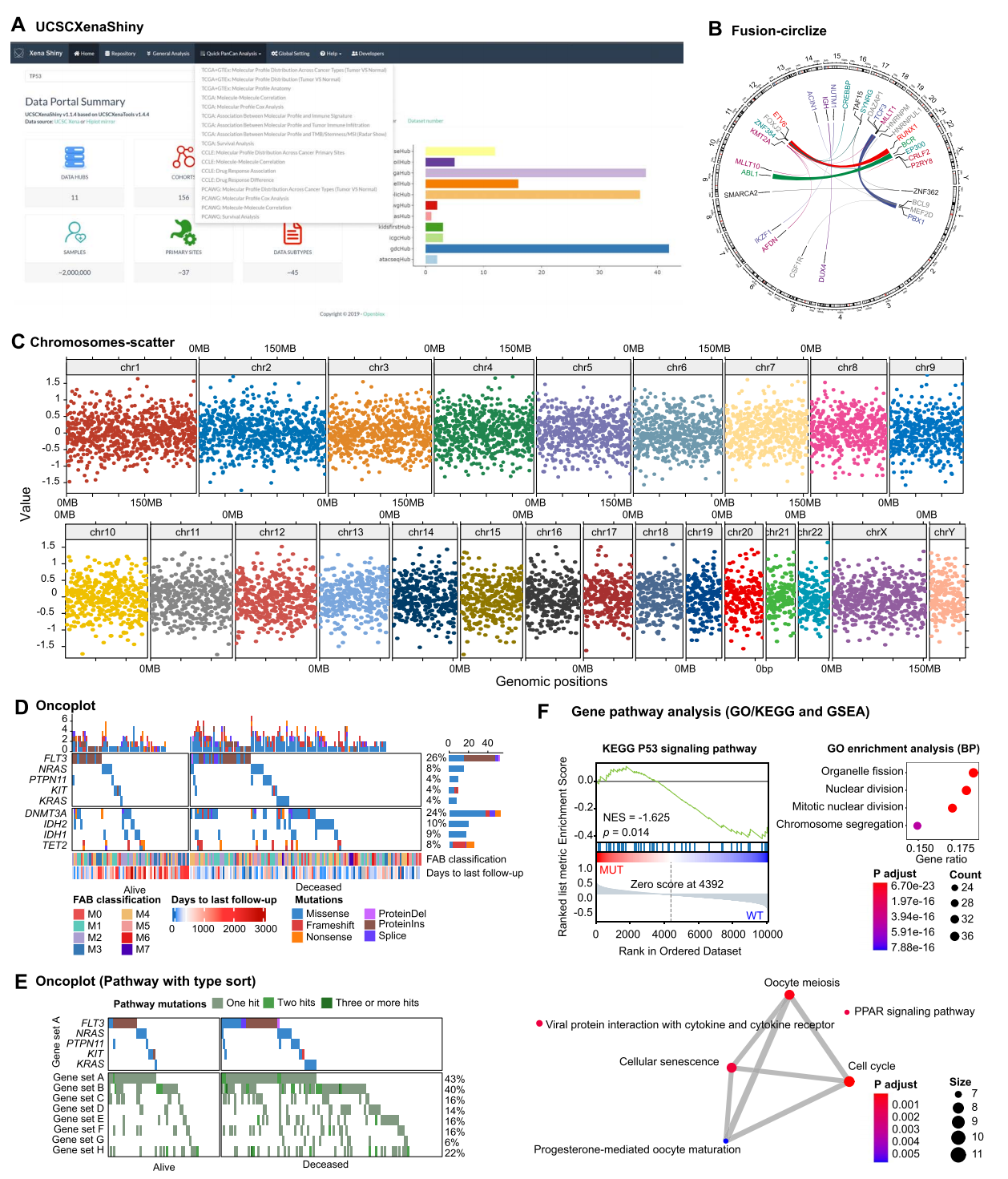
Figure 4. Hiplot 中基于组学的可视化功能的代表性用例
(A) 图为 UCSCXenaShiny 应用程序界面截图,该基于 R Shiny 开发的应用可交互式挖掘 UCSC Xena Hub 的公开数据集。
(B) fusion-circlize 插件的示例输出。展示了 B 细胞前体急性淋巴细胞白血病 (BCP-ALL) 中已知的基因融合事件,包括 ETV6::RUNX1、BCR::ABL1、DUX4 fusions、ZNF384 fusions、MEF2D fusions 和 KMT2A fusions 等。染色体按顺时针方向排列,基因融合事件通过彩带连接不同染色体,连接线及文本的颜色与宽度分别代表融合基因类别和出现频率。
(C) chromosomes-scatter 插件的示例输出。不同颜色表示各染色体上的模拟数值分布。
(D) Oncoplot 展示 TCGA 数据库中急性髓系白血病队列的选定突变基因,患者按生存状态分为两组,不同突变类型以颜色区分,底部方框显示患者元注释信息。
(E) 另一个 Oncoplot 通过添加基因通路行和患者数据实现突变分类可视化。
(F) gsea 和 clusterprofile-go-kegg 插件的示例输出:GSEA 结果以 PDF 格式呈现,显示标准化富集分数(NES)和 P 值;clusterprofile-go-kegg 插件则通过气泡图和网络图展示基于示例数据的 GO 通路富集分析结果。
除基于 Shiny 的应用程序外,Hiplot 平台还提供多个基于 JSON 的原生插件用于交互式可视化大规模癌症组学数据。例如:融合基因环状图插件 (fusion-circlize) 在染色体层面展示 B 细胞前体急性淋巴细胞白血病 (BCP-ALL) 中的典型基因融合事件,包括 RhoGEF 与 GTPase (BCR)-ABL proto-oncogene 1、ETS 变异转录因子 6 (ETV6)-RUNX 家族转录因子 1 (RUNX1)、双同源框基因 4 (DUX4) 融合、锌指蛋白 384 (ZNF384)融合、肌细胞增强因子2D(MEF2D)融合、组氨酸甲基转移酶2A(KMT2A)融合以及睾丸核蛋白(NUTM1)融合等 (Figure 4B)。染色体散点图插件 (chromosomes-scatter) 通过染色体坐标散点展示数值分布 (Figure 4C),可用于全基因组尺度下基因拷贝数和表达水平的比较分析。而肿瘤突变图谱插件 (oncoplot) 则可视化 TCGA 急性髓系白血病(LAML)队列中患者特异性突变基因与通路信息 (Figure 4D and E)。discover-mut-test 插件通过互斥共现分析算法,在识别基因互斥事件方面表现出优于传统 Fisher 精确检验的性能。基于已发表的 BCP-ALL 患者变异数据,作者使用多个 Hiplot 插件证实 BCP-ALL 中主要融合基因与染色体异常多呈互斥关系,但细胞因子受体样因子2(CRLF2)融合、DUX4融合和BCR-ABL1突变会与Janus激酶2(JAK2)、碱性螺旋-环-螺旋(bHLH)转录因子原癌基因(MYC)及RUNX1序列变异共存 (Figure S11)。
转录组数据可通过基础与高级插件(如热图、一致性聚类、复合热图、箱线图、火山图和伪增强MA图等)进行无监督聚类和表达水平可视化。一致性聚类插件 (cola) 基于基因表达数据完成共识聚类演示 (Figure S12)。该插件通过数据采样整合多种高变异基因筛选与聚类方法,提供获取稳定分群的接口。复合热图插件 (complex-heatmap) 则可关联基因表达、基因突变与临床特征等多组学数据 (Figure S13)。获得亚群差异表达基因 (DEGs) 后,用户可在火山图中交互式添加目标基因标签。
clusterprofiler-go-kegg 插件支持基于多列数据一次性完成多组通路富集分析 (Figure 4F),可接收基因符号、Ensembl ID 等多种标识符输入,并将多图整合输出为 PDF 和 Excel 文件。基因集富集分析 (GSEA) 插件完整复现了 Broad 研究所命令行版本的功能,相比桌面版更支持同时比较多个亚群与基因集 (Figure 4G)。此外,作者重构了 immunedeconv R 包的网页界面,支持基于多种算法进行肿瘤微环境分析和免疫细胞组分计算 (Figure S14)。最后,作者采用主成分分析、t-SNE、UMAP 和自组织映射等降维方法可视化经典鸢尾花数据集,展示数值特征在降维空间中的已知分类分布 (Figure S15)。这些方法可根据特征排序或距离度量从多维组学数据中筛选重要维度作为代表性数据,有助于识别具有生物学/临床意义的潜在新细胞/患者亚型。
使用案例3:临床模型
临床模型可视化功能可用于识别疾病风险因素并构建回归模型的典型图表。本节提供五个应用案例。首先,作者使用 ezcox 插件在临床数据可视化任务中执行 Cox 回归分析,构建包含控制变量的多变量生存风险模型(Figure 5A and B);metawho 插件是"治疗获益人群 Meta 分析方法"的简易网页实现,用于开展 Meta 分析任务(Figure 5C);risk-plot 插件(Figure 5D)和 survival 插件(Figure 5E)分别用于可视化患者疾病风险模型与生存数据。risk-plot 插件展示生存状态与风险因素相关性,并按风险评分从低到高排序患者;最后,作者基于 survival 包中的肺癌数据集,通过 nomogram 插件(Figure 5F)建立预测性风险评分的演示模型。
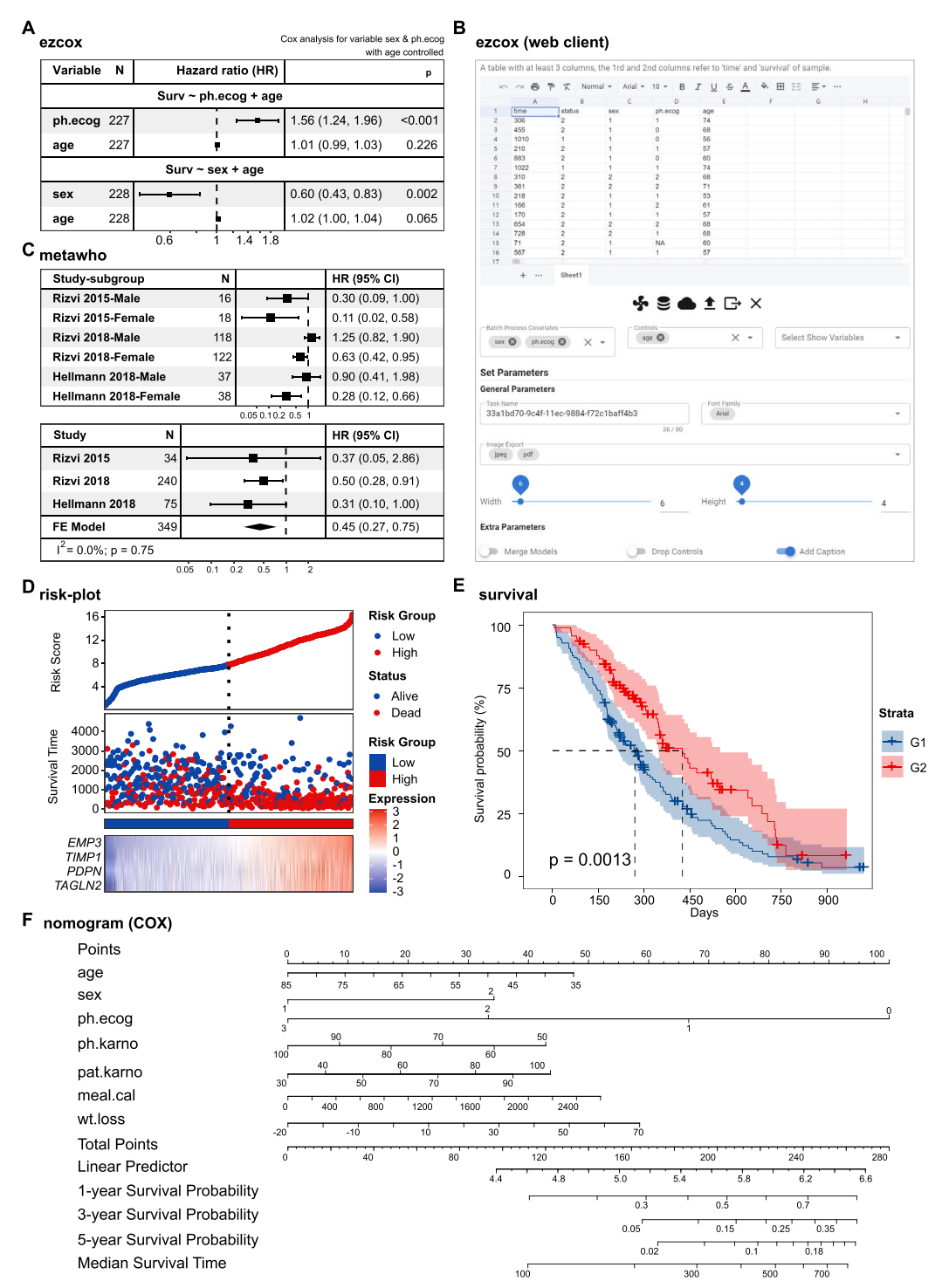
Figure 5. Hiplot 中临床数据可视化的代表性用例
(A-B) 分别展示了使用 survival 包中肺癌数据集的森林图示例及 ezcox 插件的网页界面。Cox 模型显示,当以年龄作为控制变量时,性别(sex)和 ECOG 评分(ph.ecog)分别与高风险(HR: 1.56, CI: 1.24–1.96)和低风险(HR: 0.60, CI: 0.43–0.83)显著相关。
(C) metawho 插件的示例输出,表格中展示了各亚组研究和整体研究的风险比(HR)。
(D) risk-plot 插件的示例结果。根据风险评分中位数将患者分为高风险(红色)和低风险(蓝色)亚组,每个点代表一名患者。中间区域红色点表示死亡患者,底部区域则显示特征基因表达水平,包括上皮膜蛋白3(EMP3)、金属蛋白酶组织抑制剂1(TIMP1)、足萼蛋白(PDPN)和转胶蛋白2(TAGLN2)。
(E) survival 插件的模拟数据输出,展示 3 年生存曲线。
(F) 基于 survival 包肺癌数据集构建的列线图(nomogram)预测模型,P 值通过对数秩检验计算得出。HR:风险比;CI:置信区间。
3. 用户任务和基准测试显示基于本机 Hiplot 插件的性能
作者收集了 2022 年 4 月 18 日至 24 日期间基础工具、高级工具和迷你工具插件的运行耗时数据,并进行了可视化基准测试,为后续可能的优化提供参考。数据显示,基础模块和高级模块中分别有超过 80% 的插件可在 5 秒内和 3 分钟内完成分析(Figure 6A)。此外,用户提交的任务表明,大多数用户倾向于上传小于 10MB 的文件至本网站进行可视化分析(Figure 6B)。在耗时超过 1 分钟的插件中(至少 10 次任务记录),世界地图插件的中位耗时最长,而热图和相关性图则是使用频率最高的两个插件(Figure 6C)。
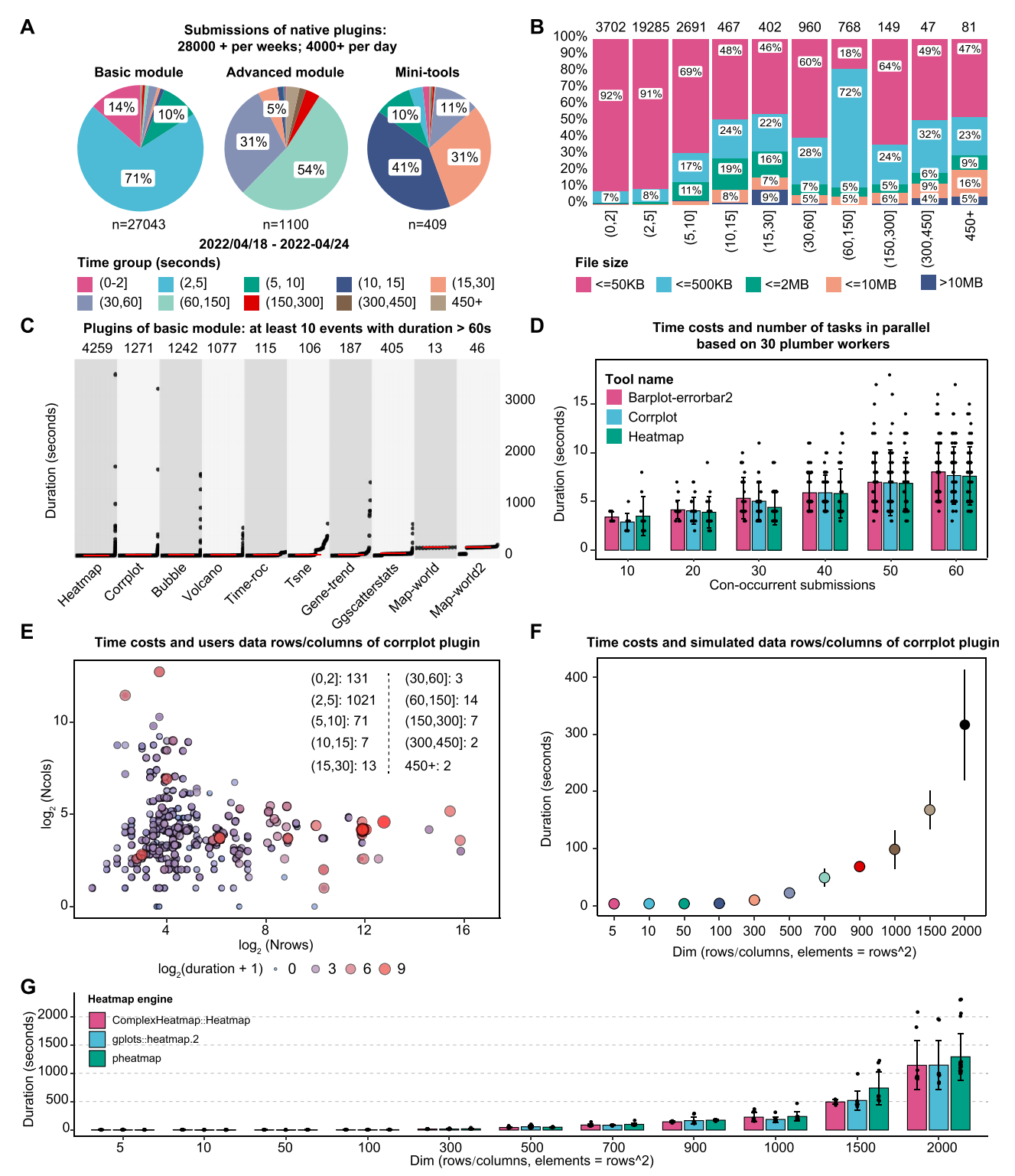
Figure 6. 用户任务和选定本机插件的基准测试显示了 Hiplot 的性能
(A) 饼图展示 2022 年 4 月 18 日至 24 日一周内基础工具、高级工具和迷你工具插件的耗时分布,不同颜色代表不同耗时区间组别。
(B) 条形图显示任务数据量与耗时的关系,不同颜色表示不同文件大小分组。
(C) 散点图呈现基础模块中耗时超过 60 秒且执行次数 ≥10 次的任务,插件按中位耗时从低(左)到高(右)排序,面板顶部标注总提交次数。
(D) 带误差点的条形图说明并行提交对任务耗时的影响,barplot-errorbar2、corrplot 和 heatmap 插件用不同颜色区分。
(E) 气泡图展示任务耗时与数据行列数的正相关关系。
(F) 误差棒散点图显示 corrplot 插件在不同数据维度下的耗时表现。
(G) 条形图比较不同热图引擎处理不同维度数据时的耗时差异。
作者进一步通过调用并行任务进行基准测试。结果显示,热图(heatmap)、相关性图(corrplot)和误差棒条形图(barplot-errorbar2)等插件的并行任务数量与耗时呈正相关。不过,在 10 至 60 个并行提交的不同测试条件下,大多数任务仍能在 15 秒内完成(当前 Hiplot 实际并发数小于 10)(Figure 6D)。数据行列数是影响耗时的另一关键因素。以相关性图插件为例,虽然大部分提交可在 5 秒内完成,但行列数增加会显著延长处理时间(Figure 6E)。基于不同维度数据矩阵的标准测试表明,当数据超过 1000 行/列(即 100 万个元素)时,相关性图插件的性能开始下降(Figure 6F)。采用不同 R 语言热图引擎实现的相关性热图插件也呈现相似规律(Figure 6G)。
4. 用户访问代表受欢迎程度和潜在影响
Hiplot 项目于 2019 年启动,首个正式版本于 2021 年 3 月发布。作者对 2020 年 7 月 9 日至 2021 年 12 月 31 日的网站访问数据统计显示:该平台网页端已获得来自全球 100 个国家超 250 万次访问,日均访问量超 5000 次,日均任务提交量达 3000 次(Figure S16A)。平台注册用户数突破 2.2万(Figure S16B),其中 160 余个插件访问量逾千次(Figure S16C)。基础热图插件单模块访问量超 7 万次,相关性热图、气泡图、箱线图、线性回归和火山图等核心插件访问量均突破 2 万次。基于 clusterProfiler 和 GSEA 的通路分析工具分别实现 2.1 万次和 5000 次调用,高级模块中的 UCSCXenaShiny 应用访问量达 6300 次,迷你工具模块的 PDF 拼图插件使用超 7000 次。这些流量增长数据印证了平台部署的各类网络插件所产生的学术影响力。
讨论
当今生物学/生物医学与计算科学的联系比数十年前更为紧密。多维数据可视化技术(如现代统计图表和组学数据可视化)已成为生物医学数据挖掘不可或缺的工具。目前研究人员主要通过三种途径实现生物医学数据可视化:传统商业桌面软件、编程语言/库以及网络工具。然而,绘制高质量、可直接出版的科学图表仍是生物信息学家和专业数据分析师面临的挑战,他们通常需借助 R、Python 等编程语言库来完成,这对缺乏编程技能的生物学家和临床医生更为困难。因此,基于现代网络技术的生物医学可视化工具正日益受到科研界青睐。
现有基于云计算的平台主要聚焦上游组学分析流程,对轻量级科学数据可视化和交互式分析的支持不足,且缺乏实时更新能力。这些平台往往操作复杂,在处理大数据时步骤冗余,其网页插件开发模式难以友好支持基于表格的轻量级可视化任务。科研用户对现代科学图表系统实现和交互式数据挖掘应用的高频需求,对推动生物医学领域数据科学发展具有重要意义。
本研究提出的 Hiplot 云服务平台,为生物医学数据交互式可视化提供了全面易用的解决方案。其简洁的用户界面(如基于电子表格的数据导入)和高效交互设计,极大降低了非编程用户的轻量级可视化任务学习成本。作者通过支持版本控制的多用户 Plumber 服务器和基于 JSON 的结构化插件系统,加速了轻量级科学可视化插件的开发。自 2019 年以来,Hiplot 联盟已实现数百个生物医学可视化插件。本研究不仅提供了该综合平台的用户使用全景数据,还通过精选原生插件进行基准测试,这些工作将促进 Hiplot 及相关工具的迭代优化。尽管该网站在处理大数据和上游组学分析方面仍有改进空间,但现有可视化插件已为生物医学数据挖掘提供了重要在线资源。未来平台将扩展更多可视化功能和公共生物医学数据集的交互式挖掘。这一新兴工具包将持续惠及缺乏编程技能的生物信息学、生物医学及其他领域的数据科学家。
--------------- 结束 ---------------
注:本文为个人学习笔记,仅供大家参考学习,不得用于任何商业目的。如有侵权,请联系作者删除。
