单细胞学习(12)——RunUMAP() 详解学习笔记
R 语言 RunUMAP() 详解
UMAP(Uniform Manifold Approximation and Projection)是一种流行的非线性降维方法,特别适用于单细胞 RNA 测序(scRNA-seq)等高维数据可视化。而在 Seurat 包中,我们主要通过 RunUMAP() 来执行 UMAP 计算。
这篇学习笔记记录 RunUMAP() 的用法,搞清楚它的关键参数,并提供一些调优技巧,让UMAP 结果更加美观、稳定。
1. RunUMAP() 的基本用法
在 Seurat 分析流程中,RunUMAP() 一般用于 PCA 降维之后。代码示例如下:
data_object <- RunUMAP(data_object, dims = 1:30, n.neighbors = 20, min.dist = 0.3)
这是最常见的 UMAP 运行方式,默认基于 PCA 降维后的数据进行计算。别急,我们来逐个拆解参数。
2. RunUMAP() 的核心参数解析
| 参数 | 默认值 | 作用 |
|---|---|---|
reduction = "pca" | "pca" | 指定 UMAP 运行的降维数据来源,一般是 PCA 结果。 |
dims = 1:50 | 1:50 | 选择 PCA 维度(通常设 30 左右)。 |
n.neighbors = 30 | 30 | UMAP 计算的邻居点数量,决定局部 vs 全局结构。 |
min.dist = 0.3 | 0.3 | 控制点的聚合程度,越小点越紧密,越大点越松散。 |
spread = 1 | 1 | 影响 UMAP 低维表示的整体扩展程度。 |
metric = "cosine" | "cosine" | 计算相似度的度量方式,默认使用余弦相似度。 |
n.components = 2 | 2 | UMAP 的降维维度,通常设为 2D(可设 3D)。 |
seed.use = 42 | 42 | 设置随机种子,以保证 UMAP 结果可复现。 |
其中,n.neighbors 和 min.dist 是影响 UMAP 结果最关键的两个参数,调整得当可以让 UMAP 结果更符合你的需求。
3. RunUMAP() 运行过程解析
当你运行 RunUMAP() 时,Seurat 内部执行以下计算步骤:
- 降维预处理:选择 PCA 结果(默认 30 维)。
- 邻近点搜索(KNN 计算):使用 Annoy 计算最近邻(
n.neighbors = 30)。 - 距离校准:平滑化 KNN 结果。
- UMAP 初始化:使用拉普拉斯归一化生成初始嵌入。
- UMAP 优化:运行 200 轮优化,将高维数据投影到 2D/3D 空间。
此外,Seurat 4.x 版本默认使用 R-native UWOT 计算 UMAP,而非 Python umap-learn,如果需要使用 Python 版本,可以手动指定:
RunUMAP(data_object, umap.method = "umap-learn", metric = "correlation")
4. 如何调整 UMAP 结果?
4.1 如果 UMAP 结果太散,看不清聚类
调整方案:
RunUMAP(data_input, dims = 1:30, n.neighbors = 15, min.dist = 0.1)
- 减少
n.neighbors:让 UMAP 更关注局部结构。 - 降低
min.dist:让点更加聚集。
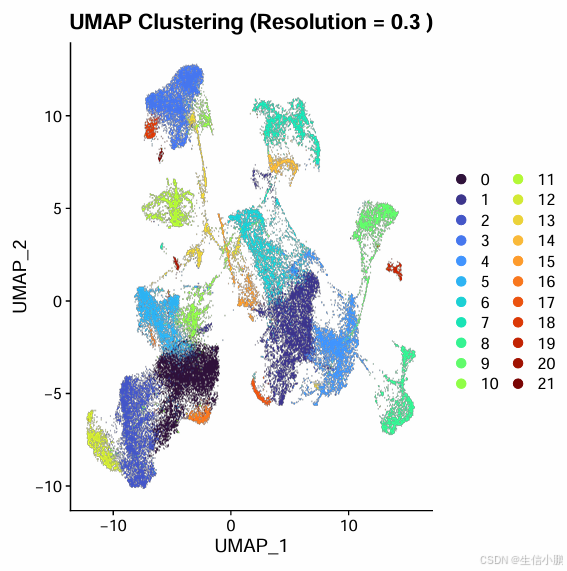
4.2 如果 UMAP 结果过于紧密,类别堆在一起
调整方案:
RunUMAP(data_input, dims = 1:30, n.neighbors = 50, min.dist = 0.5)
- 增大
n.neighbors:让 UMAP 更关注全局关系。 - 提高
min.dist:使点更加分散。
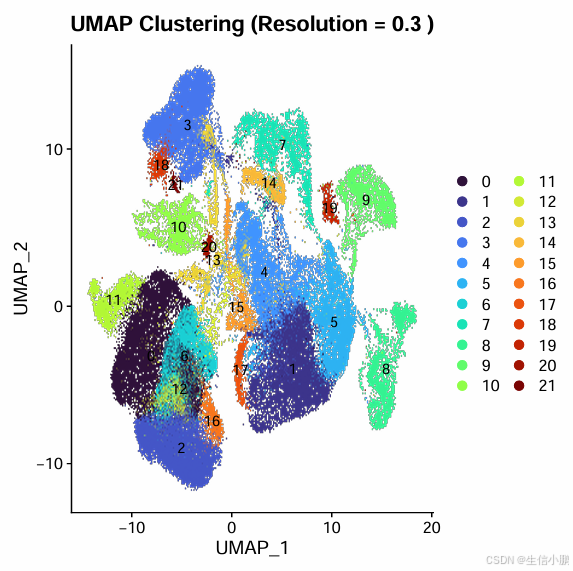
4.3 运行 3D UMAP
如果想要 3D 可视化:
RunUMAP(data_input, dims = 1:30, n.components = 3)
然后用 plotly 进行 3D 绘图。
5. FindClusters() 的参数解析
FindClusters() 是 Seurat 中用于聚类分析的函数,通常与 RunUMAP() 搭配使用。其核心参数如下:
| 参数 | 默认值 | 作用 |
|---|---|---|
resolution | 0.8 | 控制聚类的颗粒度,值越大,聚类数越多。 |
algorithm | 1 | 选择不同的聚类算法(1 = Louvain,2 = Louvain with multilevel refinement,3 = SLM)。 |
graph.name | NULL | 指定用于聚类的图(通常是 FindNeighbors() 生成的)。 |
random.seed | NULL | 设定随机种子,以确保聚类结果可复现。 |
verbose | TRUE | 是否显示计算过程的日志信息。 |
是否需要设定随机种子?
是的,FindClusters() 由于涉及到随机初始化,默认情况下,每次运行的结果可能会有所不同。如果希望聚类结果可复现,建议在运行前固定随机种子,如:
set.seed(42) # 设定全局随机种子
FindClusters(data_input, resolution = 0.8, random.seed = 42)
这样,每次运行 FindClusters() 时都会得到相同的结果,确保分析的稳定性。
6. 结语
UMAP 是 scRNA-seq 可视化的利器,而 RunUMAP() 在 Seurat 中的实现提供了丰富的参数调节空间。同时,FindClusters() 作为关键的聚类方法,与 UMAP 搭配使用,能帮助研究者更好地理解细胞群体结构。
理解 n.neighbors 和 min.dist 的作用,并学会如何调整它们,可以让 UMAP 结果更加清晰、易读。而在聚类分析中,设定 resolution 以及 random.seed 可以让聚类结果更稳定、更具解释性。
下次你在 scRNA-seq 分析时,不妨试试调整这些参数,让你的 UMAP 和聚类结果更完美!🚀