2025年深圳杯D题第二版本python代码 论文分享
法医物证鉴定中的DNA分析技术,特别是基于短串联重复序列(STR)的检测方法,已成为犯罪现场调查和身份鉴定的关键技术手段。STR基因座作为染色体上的特定位置,其核心序列重复次数的多态性形成了独特的DNA指纹。在STR图谱分析中,主峰的size和height分别代表等位基因的DNA片段长度和相对含量,通过分析个体在特定基因座上的两个等位基因组合(基因型)可实现精准身份识别。针对多人犯罪案件,混合STR图谱的解析尤为关键,需要准确识别其中各DNA组分的贡献者构成及比例,为案件侦破提供重要科学依据。该技术通过STR位点的多态性分析,为维护国家安全、公共安全和社会稳定提供了强有力的法医学支持。
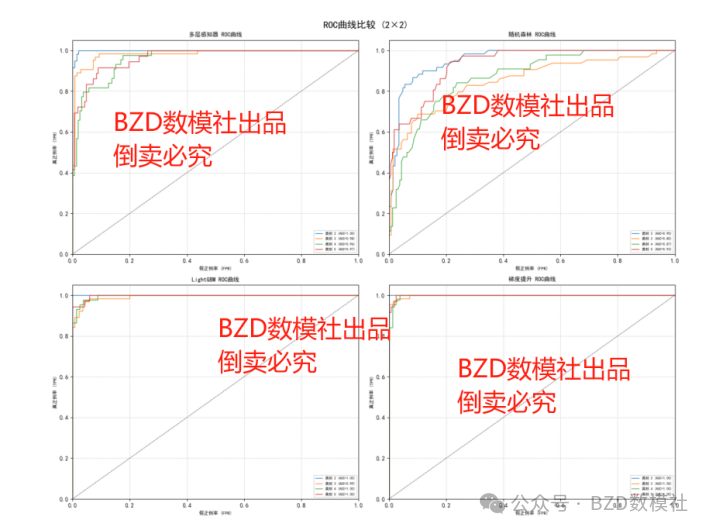
针对问题1的STR图谱贡献人数预测任务,我们首先对原始数据进行预处理,包括删除全空列、填充缺失值,并从样本文件名中提取贡献人数作为标签;接着构建包含等位基因计数、峰高统计特征和标记特异性特征的指标体系;然后将数据按比例划分为训练集和测试集并进行标准化处理;之后依次训练多层感知器、随机森林、LightGBM和梯度提升四种模型,通过准确率、精确率、召回率和F1分数等指标评估性能;最终选择在测试集上表现最优的梯度提升模型(准确率96.57%)作为解决方案。
针对问题2,提出了一种多模型融合的混合DNA样本比例识别方法。通过特征选择和标准化预处理后,采用随机森林、LightGBM、SVM和MLP四种模型进行训练,并优化模型参数防止过拟合。实验显示不同模型适用于不同复杂度的混合比例:MLP在简单比例(1:1)表现最佳(AUC=0.9302),SVM适合中等比例(1:1:1,AUC=0.7494),LightGBM则对复杂比例(1:1:1:1:1,AUC=0.6954)更具优势。
针对问题3,要求推断某一混合STR图谱中各个贡献者对应的基因型,首先通过卡方检验筛选显著STR位点特征,采用分层抽样构建训练集;建立包含岭回归、LASSO、随机森林、SVR等模型的集成系统,通过两步优化(先匹配等位基因组合,再优化峰高比例)推断贡献者基因型;最终结合兼容性分数(0.4权重)与峰高匹配分数(0.6权重)的综合评价指标选择最优模型确定最优解。
针对问题4,要求减少混合样本中噪声的干扰,首先进行数据预处理,然后采用标准小波去噪(standard_wavelet)、中值滤波去噪(median_filter)和高斯滤波去噪(gaussian_filter)三种方法进行去噪。评估结果显示高斯滤波去噪效果最优。
关键词:梯度提升;多模型融合算法;SVM;高斯滤波去噪;检验与分析;
一、问题求解与分析
4.1 问题1求解与分析
4.1.1 问题1分析
针对问题1的STR图谱贡献人数预测任务,我们首先对原始数据进行预处理,包括删除全空列、填充缺失值,并从样本文件名中提取贡献人数作为标签;接着构建包含等位基因计数、峰高统计特征和标记特异性特征的指标体系;然后将数据按比例划分为训练集和测试集并进行标准化处理;之后依次训练多层感知器、随机森林、LightGBM和梯度提升四种模型,通过准确率、精确率、召回率和F1分数等指标评估性能;最终选择在测试集上表现最优的梯度提升模型(准确率96.57%)作为解决方案,该模型在所有人数类别上均展现出稳定的预测能力。
4.1.2 问题1建模与求解
1、数据统计分析与预处理
(1)数据清洗与合并
在本次法医物证多人身份鉴定项目中,数据预处理是确保分析结果准确性的关键步骤。针对我们给出的附件的数据,首先要做的就是对数据进行预处理操作。在数据集加载后,检查每一列是否存在全为空的情况。空列没有任何有效的信息,会对后续的分析造成干扰,因此我们需要将这些列删除。由于不同的等位基因长短也不一,所以还需要对于数据中的缺失值(NaN),我们选择将其填充为0。这样的处理方式适用于数值型数据,并能避免后续计算中出现NaN值导致的错误。
由于问题1需要识别某一混合样本中的贡献者人数,为了后续对准确性进行评估,采用如下方式进行下划线分割来提取标签。
由于样本文件名的模式为RD14-8003-123_456_789-abc123,其中 123_456_789 表示贡献者ID。
提取出的贡献者ID字符串为
。然后将字符串
按照下划线分割,得到
(2)数据计数标注与标准化
针对清洗与合并后的附件1,在使用模型识别某一混合样本中的贡献者人数前,还需要做如下四步数据预处理,如表2所示。
表2
| 步骤 | 目的 | 数学基础 |
| 等位基因计数 | 量化信号复杂度 | 计数统计 |
| 高度统计特征 | 描述峰高分布 | 均值、标准差、极值比 |
| 标准化 | 消除特征量纲差异 | Z-score标准化 |
| 标记特异性特征 | 捕获不同标记的差异 | 独热编码(二进制指示) |
具体情况如下:
①等位基因技术
目的:量化样本中检测到的等位基因信号数量
统计非空等位基因(Allele)和非OL(Off-Ladder)等位基因的数量数学公式:
(3)数据统计分析
在数据预处理完成之后,我们可以开始进行数据可视化操作,以便直观地了解数据的分布特征和关系。

图1呈现了每个样本的峰值数量分布,采用箱型图展示了不同样本的峰值数量的变化情况。从图中可以看出,每个样本的峰值数量(即每个样本中的有效DNA峰值数)有所不同。大多数样本的峰值数量集中在5到15之间,少数样本的峰值数量明显较多,达到20个以上。这些较大的峰值数量可能表示样本的复杂性较高,例如样本中有多个来源的DNA(混合样本)。整体而言,样本的峰值数量分布存在一定的离散性,但大部分集中在一个相对狭窄的范围内。箱型图的中位数(橙色线)显示出该数据的中心趋势,箱体的上下边缘分别表示四分位数
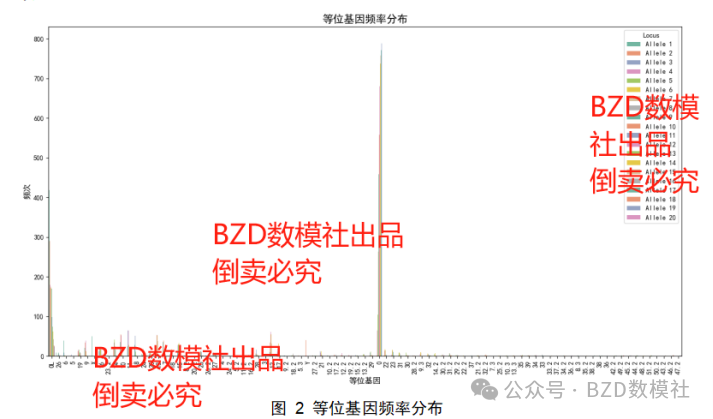
图2展示了在不同基因座上,所有等位基因的频率分布情况。从图中的颜色区分可以看到,等位基因的分布存在明显的偏倚,某些等位基因出现的频率极高(如等位基因20),而其他等位基因则出现的频率相对较低。数据点密集的区域表明某些基因座的DNA片段长度在样本中较为一致,导致这些等位基因的频率较高。这类图表有助于观察特定等位基因在样本中的普遍性,并能为后续的样本混合识别提供依据。该分布有助于法医物证的鉴定工作,能够显示出特定基因座的变异情况。
至此,数据统计分析与预处理完毕,具体的过程如图5所示。
| 模型 | 准确率(Accuracy) | 精确率(Precision) | 召回率(Recall) | F1分数 | 类别2 F1 | 类别3 F1 | 类别4 F1 | 类别5 F1 |
| 多层感知器 | 0.8873 | 0.8900 | 0.8873 | 0.8873 | 0.9677 | 0.9268 | 0.8000 | 0.7895 |
| 随机森林 | 0.7059 | 0.7028 | 0.7059 | 0.7004 | 0.7970 | 0.7087 | 0.5926 | 0.6567 |
| LightGBM | 0.9510 | 0.9510 | 0.9510 | 0.9509 | 0.9917 | 0.9291 | 0.9213 | 0.9577 |
| 梯度提升(最佳) | 0.9657 | 0.9673 | 0.9657 | 0.9659 | 1.0000 | 0.9688 | 0.9231 | 0.9565 |