案例解析:基于量子计算的分子对接-QDOCK(Quantum Docking)
分子对接(Moleculardocking)在药物发现中具有重要意义,但对接的计算速度和准确率始终难以平衡,其巨大解搜索空间对传统计算机来说异常艰巨。
本文通过引入网格点匹配(GPM, Grind point matching)和特征原子匹配(FAM, Feature atom matching)两种方法,将分子对接问题中的采样过程编码为二次无约束二进制优化(QUBO)问题来加速分子对接,使其能够通过量子计算机(如相干伊辛机CIM)进行求解。结果表明,GPM的采样能力接近Glide SP(一种进行广泛搜索的方法),并且据估计,同等问题的求解在CIM硬件上要比在传统计算机上快至少1000倍。该方法有望在未来加速小分子和多肽的药物虚拟筛选。
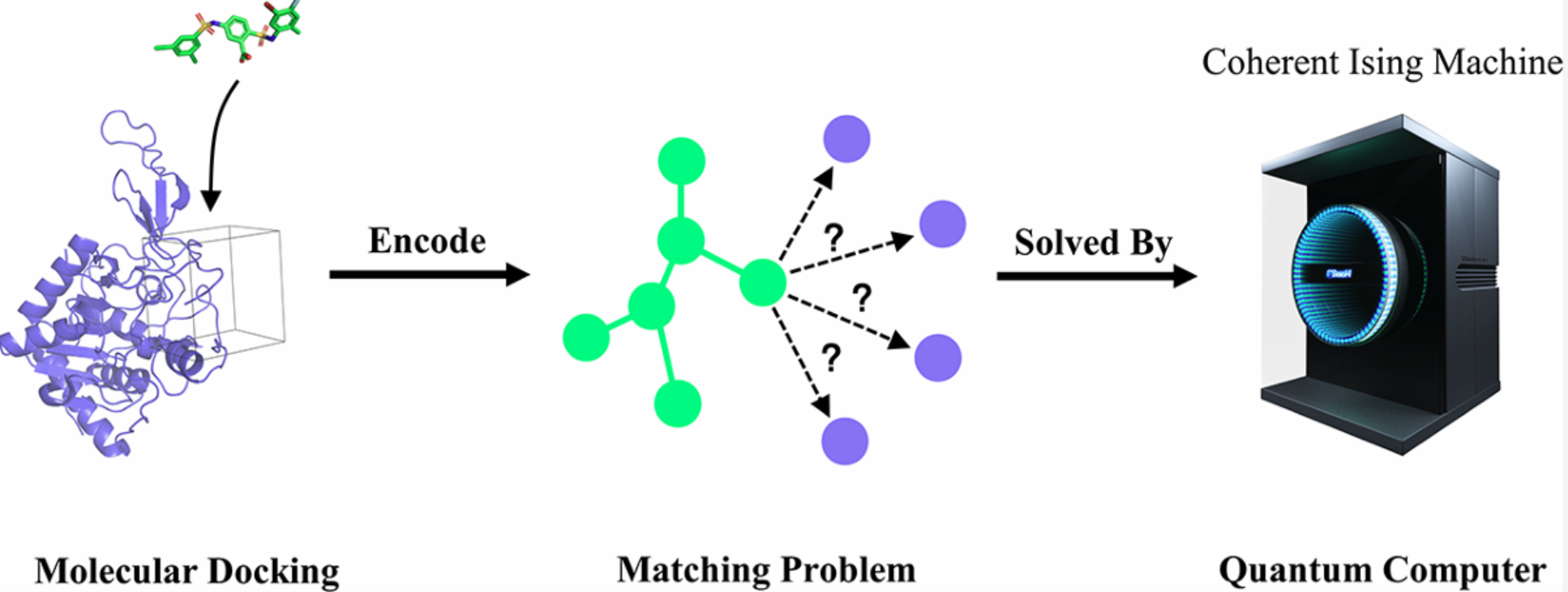 图1 基于光量子计算机的分子对接示意图
图1 基于光量子计算机的分子对接示意图
一、背景介绍
分子对接是一种广泛应用于虚拟筛选、先导化合物优化和机制研究的计算技术,其核心是通过优化配体与蛋白质的结合姿态和自由能 ( Δ G B i n d ) (\Delta G_{Bind}) (ΔGBind)确定结合模式。
然而,该问题的NP-hard特性导致精确求解需枚举大量构象,现有方法(如模拟退火、分步枚举、深度学习模型和GPU并行计算)仍难以应对十亿级数据库(如ZINC)的筛选需求。量子计算机(如量子退火器和CIM)为解决NP-hard问题提供了新途径,其通过将问题映射为QUBO模型并利用量子比特的相互作用实现高效求解。
二、建模思路
2.1 问题表述
分子对接的采样过程是寻找到一个配体分子和靶蛋白结合的具体构象,用数学形式可表示成以下模型:假设一个配体由 n n n个原子组成 ( a 1 , a 2 , a 3 , … , a n ) (a_1,a_2,a_3,\dots,a_n) (a1,a2,a3,…,an),第 i i i个原子 a i a_i ai的坐标表示为 r i r_i ri,则一个分子对接任务就是最小化以下目标函数:
Δ G Bind ( a 1 , a 2 , … , a n , r 1 , r 2 , … , r n ) r i ∈ D \Delta G_{\text{Bind}}(a_1, a_2, \dots, a_n, \mathbf{r}_1, \mathbf{r}_2, \dots, \mathbf{r}_n) \mathbf{r}_i \in \mathbf{D} ΔGBind(a1,a2,…,an,r1,r2,…,rn)ri∈D D D D代表对接定义的docking box限定的对接空间,以上这个公式也被称为“能量函数”。
2.2 QUBO建模
2.1中描述的模型是一个经典的优化问题,对于传统的优化算法难以在短时间内得到一个较优的解,本文中提出一种建模思路,首先将 D D D等距离散化成 N N N个格点 ( g 1 , g 2 , g 3 , … , g n ) (g_1,g_2,g_3,\dots,g_n) (g1,g2,g3,…,gn),如果原子 a i a_i ai匹配到格点 g s i g_{s_i} gsi上, a i a_i ai和 a j a_j aj的距离是 d i j d_{ij} dij, g s i g_{s_i} gsi和 g s j g_{s_j} gsj的距离是 D s i s j D_{s_is_j} Dsisj,此时上面的函数可以表示为:
Δ G Bind ( a 1 , a 2 , … , a n , r 1 , r 2 , … , r n ) ≈ Δ G Bind ′ ( a 1 , a 2 , … , a n , g s 1 , g s 2 , … , g s n ) = ∑ i = 1 n w a i g s i s i = 1 , 2 , … , N s . t . ∣ d i j − D s i s j ∣ ≤ c dist \begin{aligned} &\Delta G_{\text{Bind}}(a_1, a_2, \dots, a_n, \mathbf{r}_1, \mathbf{r}_2, \dots, \mathbf{r}_n) \approx \Delta G'_{\text{Bind}} \\ &(a_1, a_2, \dots, a_n, g_{s_1}, g_{s_2}, \dots, g_{s_n}) = \sum_{i=1}^{n} w_{a_i g_{s_i}} \\ &s_i = 1, 2, \dots, N \quad s.t. \ |d_{ij} - D_{s_i s_j}| \leq c_{\text{dist}} \end{aligned} ΔGBind(a1,a2,…,an,r1,r2,…,rn)≈ΔGBind′(a1,a2,…,an,gs1,gs2,…,gsn)=i=1∑nwaigsisi=1,2,…,Ns.t. ∣dij−Dsisj∣≤cdist 这里的 w a i g s i w_{a_i g_{s_i}} waigsi就是原子 a i a_i ai放在格点 g s i g_{s_i} gsi上的合适度(fitness),后面会介绍合适度怎么计算的,可以把这一项看成是 Δ G B i n d \Delta G_{Bind} ΔGBind的分子间的约束项。此外,还需要增加一些约束项,即在优化过程中须保持分子本身的结构,即分子内的约束项。
此时上面的式子就可以转化成QUBO模型, x i j x_{ij} xij是一组二值变量,代表了 a i a_i ai和 g j g_j gj是否匹配(0代表不匹配,1代表匹配),所以上面的式子就可以写成二次项形式,其中 c d i s t c_{dist} cdist为距离阈值,用于添加空间几何约束:
Δ G Bind ′ ( a 1 , a 2 , … , a n , g s 1 , g s 2 , … , g s n ) = Δ G Bind ′ ( a 1 , a 2 , … , a n , g 1 , g 2 , … , g N , x 11 , x 12 , … , x n N ) = ∑ i = 1 n ∑ j = 1 N w a i g j x i j 2 x i j = { 1 , a i matches g j 0 , a i does not match g j s . t . ∣ d i k − D j l ∣ ≤ c dist and ∑ i = 1 n x i j ≤ 1 \begin{aligned} &\Delta G'_{\text{Bind}}(a_1, a_2, \dots, a_n, g_{s_1}, g_{s_2}, \dots, g_{s_n}) \\ &= \Delta G'_{\text{Bind}}(a_1, a_2, \dots, a_n, g_1, g_2, \dots, g_N, x_{11}, x_{12}, \dots, x_{nN}) \\ &= \sum_{i=1}^{n}\sum_{j=1}^{N} w_{a_i g_j} x_{ij}^2 \\ &x_{ij} = \begin{cases} 1, & a_i \text{ matches } g_j \\ 0, & a_i \text{ does not match } g_j \end{cases} \\ &s.t. \ |d_{ik} - D_{jl}| \leq c_{\text{dist}} \text{ and } \sum_{i=1}^{n} x_{ij} \leq 1 \end{aligned} ΔGBind′(a1,a2,…,an,gs1,gs2,…,gsn)=ΔGBind′(a1,a2,…,an,g1,g2,…,gN,x11,x12,…,xnN)=i=1∑nj=1∑Nwaigjxij2xij={1,0,ai matches gjai does not match gjs.t. ∣dik−Djl∣≤cdist and i=1∑nxij≤1
加入惩罚项
但是,在优化上面这个式子时,有两点约束条件要考虑:一是保持配体形状;二是一个格点只能匹配一个原子。因此,可以将约束也写成二次多项式形式,二者的惩罚项系数分别记为 K d i s t K_{dist} Kdist(保持配体形状)和 K m o n o K_{mono} Kmono(原子和格点的一一对应),如下所示:
Δ G Bind ′ ′ ( a 1 , a 2 , … , a n , g 1 , g 2 , … , g N , x 11 , x 12 , … , x n N ) = ∑ i = 1 n ∑ j = 1 N w a i g j x i j 2 + K dist ∑ i = 1 n ∑ j = 1 N ∑ k = 1 n ∑ l = j + 1 N u i j k l x i j x k l + K mono ∑ i = 1 n ∑ j = 1 N ∑ k = 1 n ∑ l = j + 1 N v i j k l x i j x k l = ( x 11 x 12 … x n N ) T ( w a 1 g 1 K dist u 1112 + K mono v 1112 ⋯ K dist u 11 n N + K mono v 11 n N 0 w a 1 g 2 ⋯ K dist u 12 n N + K mono v 12 n N ⋮ ⋮ ⋱ ⋮ 0 0 ⋯ w a n g N ) ( x 11 x 12 … x n N ) u i j k l = { 1 , ∣ d i k − D j l ∣ > c dist 0 , ∣ d i k − D j l ∣ > c dist v i j k l = { 1 , j = l and i ≠ k 0 , otherwise x i j = { 1 , a i matches g j 0 , a i does not match g j \begin{aligned} &\Delta G''_{\text{Bind}}(a_1, a_2, \dots, a_n, g_1, g_2, \dots, g_N, x_{11}, x_{12}, \dots, x_{nN}) \\ &= \sum_{i=1}^{n}\sum_{j=1}^{N} w_{a_i g_j} x_{ij}^2 + K_{\text{dist}} \sum_{i=1}^{n}\sum_{j=1}^{N}\sum_{k=1}^{n}\sum_{l=j + 1}^{N} u_{ijkl}x_{ij}x_{kl} \\ & \quad + K_{\text{mono}} \sum_{i=1}^{n}\sum_{j=1}^{N}\sum_{k=1}^{n}\sum_{l=j + 1}^{N} v_{ijkl}x_{ij}x_{kl} \\ &= \begin{pmatrix} x_{11} \\ x_{12} \\ \dots \\ x_{nN} \end{pmatrix}^T \begin{pmatrix} w_{a_1g_1} & K_{\text{dist}}u_{1112} + K_{\text{mono}}v_{1112} & \cdots & K_{\text{dist}}u_{11nN} + K_{\text{mono}}v_{11nN} \\ 0 & w_{a_1g_2} & \cdots & K_{\text{dist}}u_{12nN} + K_{\text{mono}}v_{12nN} \\ \vdots & \vdots & \ddots & \vdots \\ 0 & 0 & \cdots & w_{a_ng_N} \end{pmatrix} \begin{pmatrix} x_{11} \\ x_{12} \\ \dots \\ x_{nN} \end{pmatrix} \\ &u_{ijkl} = \begin{cases} 1, & |d_{ik} - D_{jl}| > c_{\text{dist}} \\ 0, & |d_{ik} - D_{jl}| > c_{\text{dist}} \end{cases} \quad v_{ijkl} = \begin{cases} 1, & j = l \text{ and } i \neq k \\ 0, & \text{otherwise} \end{cases} \\ &x_{ij} = \begin{cases} 1, & a_i \text{ matches } g_j \\ 0, & a_i \text{ does not match } g_j \end{cases} \end{aligned} ΔGBind′′(a1,a2,…,an,g1,g2,…,gN,x11,x12,…,xnN)=i=1∑nj=1∑Nwaigjxij2+Kdisti=1∑nj=1∑Nk=1∑nl=j+1∑Nuijklxijxkl+Kmonoi=1∑nj=1∑Nk=1∑nl=j+1∑Nvijklxijxkl= x11x12…xnN T wa1g10⋮0Kdistu1112+Kmonov1112wa1g2⋮0⋯⋯⋱⋯Kdistu11nN+Kmonov11nNKdistu12nN+Kmonov12nN⋮wangN x11x12…xnN uijkl={1,0,∣dik−Djl∣>cdist∣dik−Djl∣>cdistvijkl={1,0,j=l and i=kotherwisexij={1,0,ai matches gjai does not match gj 接下来,就可以使用量子算法对这个QUBO model进行求解,这样就可以得到在约束条件下,使得能量最低状态的原子与网格匹配的状态,即QUBO模型的解,该解可用于得到最终的采样构象。
2.3 获得配体的对接构象(docking pose)
得到原子在网格中的位置后,就可以计算RMSD (Root Mean Square Deviation),在这里只考虑刚性对接的情况,计算Kabsch RMSD旋转矩阵:
K rot = arg min K [ K ( r 1 st r 2 st … r n st ) + b − ( R s 1 R s 2 … R s n ) ] , where b = ∑ i = 1 n R s i n − ∑ i = 1 n R i st n \begin{aligned} K_{\text{rot}} &= \arg \min_{K} \left[ K \begin{pmatrix} \mathbf{r}_1^{\text{st}} \\ \mathbf{r}_2^{\text{st}} \\ \dots \\ \mathbf{r}_n^{\text{st}} \end{pmatrix} + \mathbf{b} - \begin{pmatrix} \mathbf{R}_{s_1} \\ \mathbf{R}_{s_2} \\ \dots \\ \mathbf{R}_{s_n} \end{pmatrix} \right], \text{ where } \mathbf{b} \\ &= \frac{\sum_{i=1}^{n} \mathbf{R}_{s_i}}{n} - \frac{\sum_{i=1}^{n} \mathbf{R}_{i}^{\text{st}}}{n} \end{aligned} Krot=argKmin K r1str2st…rnst +b− Rs1Rs2…Rsn , where b=n∑i=1nRsi−n∑i=1nRist 最终的对接姿态可以通过如下公式计算(这一过程可以通过Prody Python包实现):
( r 1 dock r 2 dock ⋯ r n dock ) = K rot ( r 1 st r 2 st ⋯ r n st ) + b \begin{pmatrix} \mathbf{r}_1^{\text{dock}} \\ \mathbf{r}_2^{\text{dock}} \\ \cdots \\ \mathbf{r}_n^{\text{dock}} \end{pmatrix} = K_{\text{rot}} \begin{pmatrix} \mathbf{r}_1^{\text{st}} \\ \mathbf{r}_2^{\text{st}} \\ \cdots \\ \mathbf{r}_n^{\text{st}} \end{pmatrix} + \mathbf{b} r1dockr2dock⋯rndock =Krot r1str2st⋯rnst +b
2.4 适配度 w w w的计算
为了刻画 w w w(即原子在网格点中适配度),本文引入了两种编码策略:网格点匹配(Grid Point Matching,GPM) 和 特征原子匹配(Feature Atom Matching,FAM)。这两种方法的核心思想是通过不同的方式定义配体原子 a i a_i ai与空间位置(网格点或特征原子) g j g_j gj之间的相互作用强度 w a i g j w_{a_ig_j} waigj。以下是这两种方法的具体描述:
网格点匹配(Grid Point Matching,GPM)
在GPM方法中,网格点直接在预定义的对接盒 D D D中以 2 A ˚ 2Å 2A˚的间隔生成。配体原子 a i a_i ai与网格点 g j g_j gj之间的相互作用强度 w a i g j w_{a_ig_j} waigj被定义为当原子类型 a i a_i ai放置在网格点 g j g_j gj上时的范德华力(van der Waals energy)。具体步骤如下:
- 生成网格点:在对接盒 D D D中以 2 A ˚ 2Å 2A˚的间隔生成网格点;
- 计算范德华力:使用AutoDockFR中的AutoGrid工具预计算每个网格点 g j g_j gj上的范德华力,并将其保存在每个网格点上;
- 定义 w a i g j w_{a_ig_j} waigj:对于每个配体原子 a i a_i ai和网格点 g j g_j gj, w a i g j w_{a_ig_j} waigj被定义为当原子 a i a_i ai放置在网格点 g j g_j gj上时的范德华力。
虽然AutoDockFR的打分函数中还包括静电和溶剂化项,但在GPM方法中,这些项被忽略,因为它们在离散空间中可能导致误差累积,从而影响性能。
特征原子匹配(Feature Atom Matching,FAM)
在FAM方法中,网格点首先在预定义的对接盒 D D D中以 1 A ˚ 1Å 1A˚的间隔生成,然后通过AutoSite算法将这些网格点粗粒化为3种类型的“特征原子”(Feature Atoms,FAs)。这三种特征原子分别是:
- 中性的碳原子(neutral C)
- 氢键供体氢原子(H-bond-donor H)
- 氢键受体氧原子(H-bond-acceptor O)
配体原子 a i a_i ai与特征原子 g j g_j gj之间的相互作用强度 w a i g j w_{a_ig_j} waigj被定义为配体原子和特征原子的电负性(Pauling’s electronegativity)之间的差异,具体公式为: w a i g j = ∣ χ a i − χ g j ∣ − 0.5 w_{a_ig_j} = |\chi_{a_i} - \chi_{g_j}| - 0.5 waigj=∣χai−χgj∣−0.5
。其中, χ a i \chi_{a_i} χai和 χ g j \chi_{g_j} χgj分别是配体原子 a i a_i ai和特征原子 g j g_j gj的电负性,这种定义方式反映了配体原子与特征原子之间形成氢键的倾向。
2.5 经典计算机的构象打分
之前已经提到,GPM和FAM的打分函数( Δ G Bind ′ \Delta G'_{\text{Bind}} ΔGBind′)是对 Δ G Bind \Delta G_{\text{Bind}} ΔGBind的粗略估计。实际上,本文发现【惩罚项节中的方程】在构象排序方面表现不佳。因此,本文将基于量子计算机的分子对接分为构象采样和打分两步,构象采样用于采样对接构象的可能状态,而后续的打分则用于对这些构象进行排序。相比之下,构象采样是NP-hard的,而构象打分则不是,而且它只需要一个粗略的打分函数。因此,在本文提出的的工作流程中,GPM和FAM用于构象采样,而构象打分可以留给经典计算机完成。
文章整体的思路可以由下图概括:
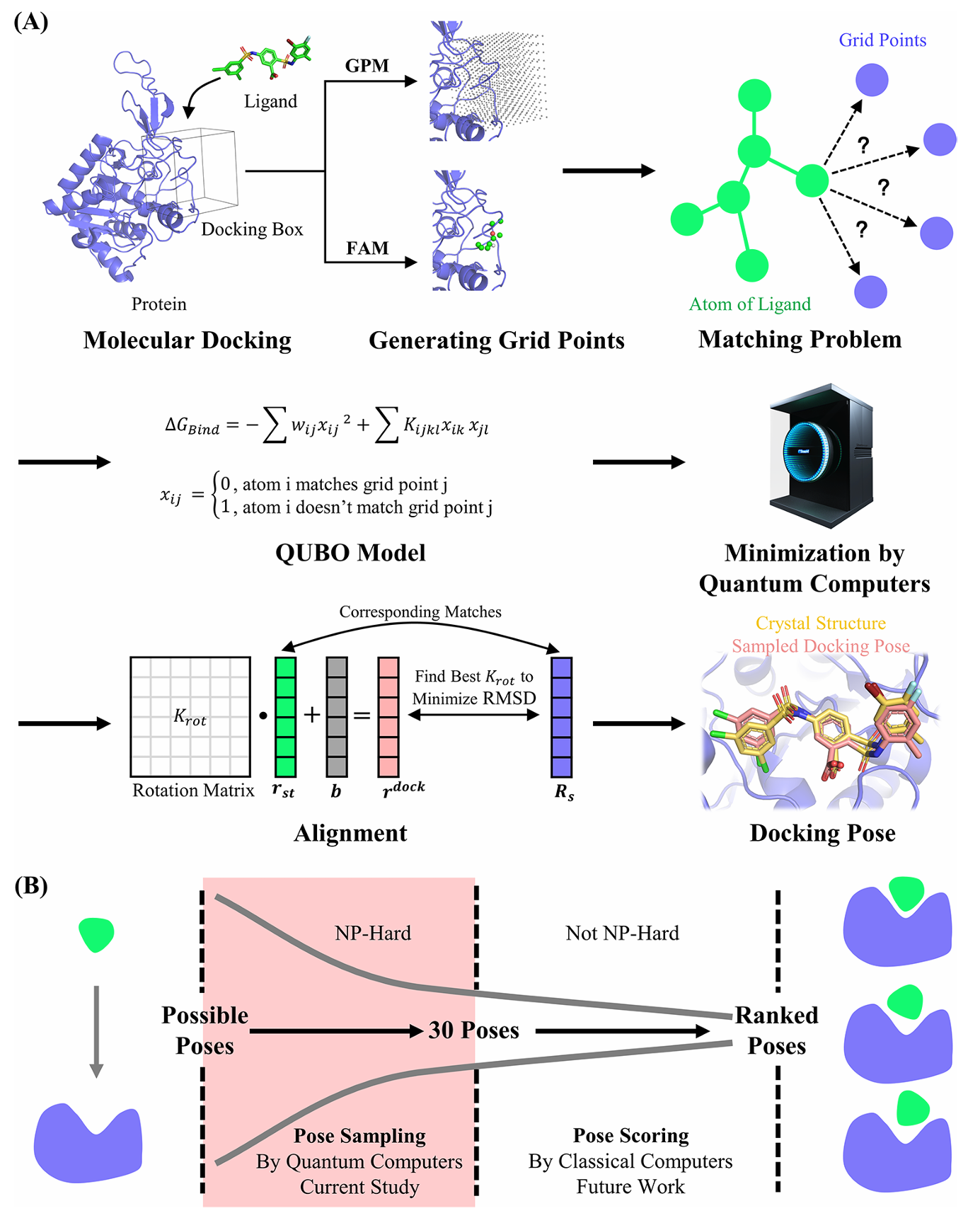 图2 基于QUBO的分子对接算法框架示意图
图2 基于QUBO的分子对接算法框架示意图
三、求解结果
3.1 采样性能比较
本文在CASF-2016数据集上评估了不同采样方法得到的构象与真实构象之间的最小差异(mRMSD, minimum RMSD),结果如下:
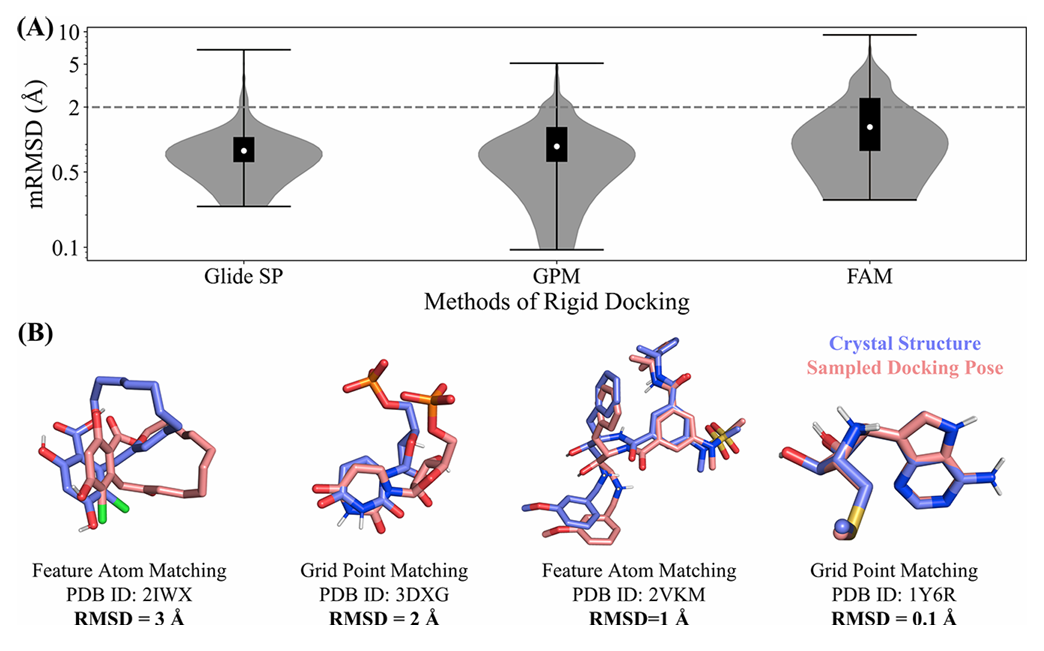 图3 GPM和FAM在CASF-2016上的采样表现
图3 GPM和FAM在CASF-2016上的采样表现
- GPM: 在257个测试案例中,GPM能够在225个案例(87.5%)中采样到高质量的对接构象(最小均方根距离mRMSD < 2 Å),平均mRMSD为1.1 Å,最大mRMSD约为5 Å。这表明GPM具有较强的采样能力,能够有效地找到接近真实晶体结构的对接构象;
- FAM: 在相同的测试案例中,FAM在173个案例(67.3%)中采样到高质量的对接构象,平均mRMSD为1.8 Å,最大mRMSD为9.4 Å。虽然FAM的性能略逊于GPM,但仍然显示出一定的有效性;
- Glide SP(benchmark): 作为一种广泛使用的对接软件,Glide SP在CASF-2016数据集的240个案例(93.4%)中能够采样到高质量的对接构象,平均mRMSD为1.0 Å,最大mRMSD为6.8 Å。GPM的性能与Glide SP接近,表明GPM在采样能力上具有竞争力。
3.2 影响采样性能的因素
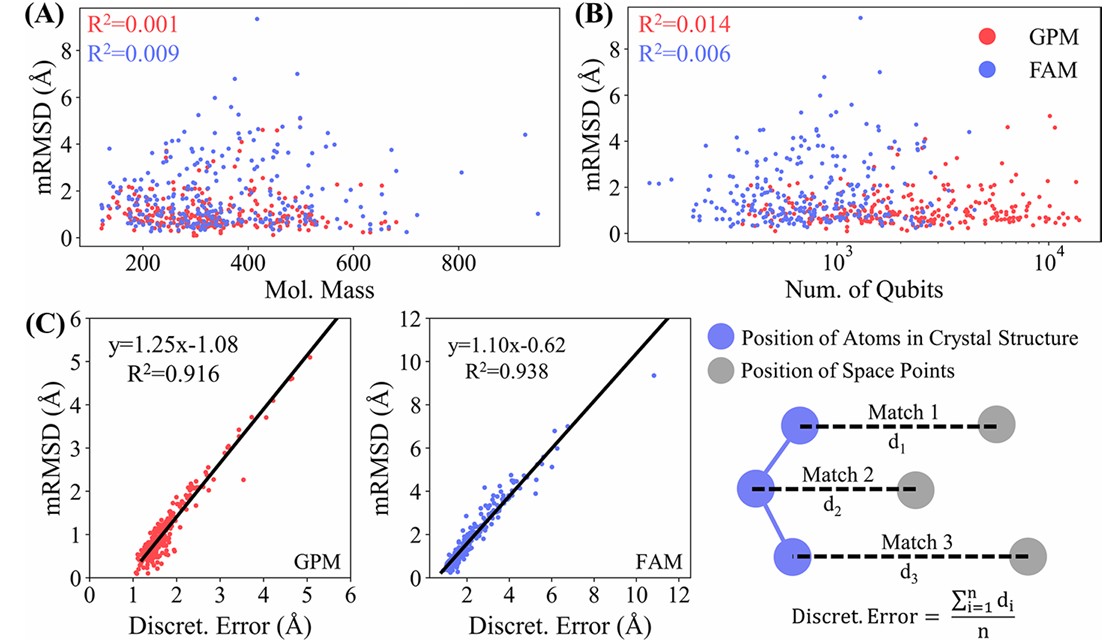 图4 GPM和FAM的采样影响因素分析
图4 GPM和FAM的采样影响因素分析
本文对可能影响采样性能的因素进行了研究,比较了如下几种因素:
- 配体质量: 采样中的mRMSD与配体质量几乎没有相关性(GPM的R² = 0.001,FAM的R² = 0.009),说明这两种方法不受配体大小的显著影响;
- 量子比特数量: mRMSD与量子比特数量的相关性也很低(GPM的R² = 0.014,FAM的R² = 0.006),表明这两种方法在量子计算资源上的效率较高;
- 离散化误差: mRMSD与离散化误差具有良好的线性关系(GPM的R² = 0.938,FAM的R² = 0.916),且回归系数接近1。这表明对接盒的离散化程度是影响采样性能的主要因素。
3.3 计算成本分析
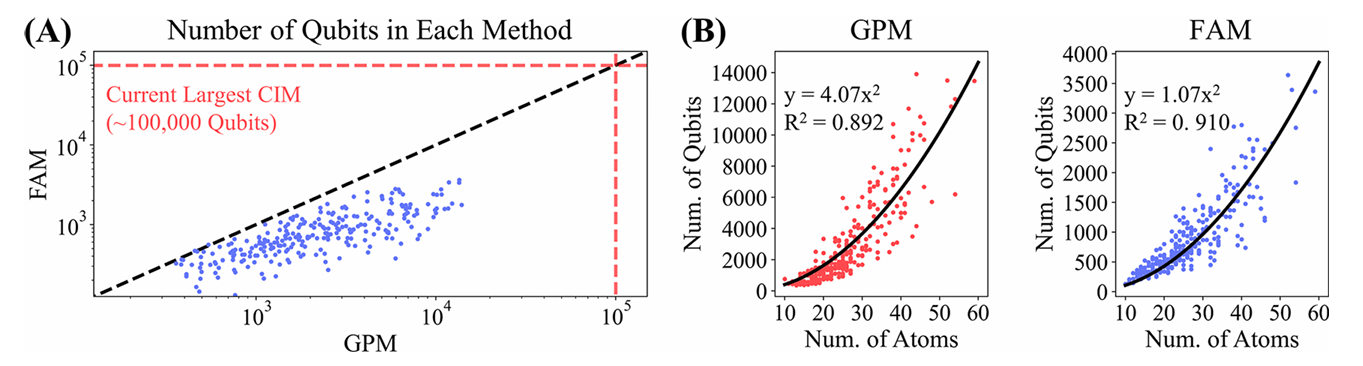 图6 GPM和FAM所需比特数分析
图6 GPM和FAM所需比特数分析
- 量子比特需求: GPM在CASF数据集上最大的量子比特需求为13,908,而FAM为3,640。这比当前实验CIM的最大量子比特数(约10万个)小1-2个数量级,为未来开发更复杂的对接方法提供了空间;
- 运行时间: 基于CIM的计算优势,采样时间估计为毫秒级,比经典计算机快3个数量级;
- 问题规模分析: GPM和FAM适用于小分子和肽的对接,因为它们的量子比特需求与配体原子数量呈二次关系,限制了输入配体原子的数量约为156(GPM)和305(FAM),分别对应于15和30个残基的肽。由于量子比特数量的限制,这两种方法目前不适用于蛋白质-蛋白质对接。
四、总结
大规模的组合优化问题求解对于传统计算硬件提出了不小的挑战,很多从算法层优化尝试在这类复杂问题上寻求一个兼顾速度和求解质量的方法。计算机辅助药物设计涉及大量的组合优化场景,在上述瓶颈下,很多问题难以在短时间内得到一个高质量的解,这种欠优的策略在药物发现领域往往失之毫厘,差之千里,造成了药物发现中先导化合物筛选假阳性率较高的境地,因此开发又快又好的计算方法是目前该领域的研究热点。
近期,量子计算硬件的发展使得在解决这类复杂问题上提出了新的解决思路,通过借助量子计算强大的并行处理能力,可全局搜所解空间,而得到能量最低的状态组合。该工作通过将分子对接过程的采样问题编码成QUBO model,可以适配光量子计算机进行求解,为分子筛选提供了全新的解决思路。
参考材料
- Zha J, Su J, Li T, et al. Encoding molecular docking for quantum computers[J]. Journal of Chemical Theory and Computation, 2023, 19(24): 9018-9024.
- 本项目中的代码目前已开源,可参考:GitHub - JinyinZha/QDock: QDock is a method encoding pose sampling in molecular docking for quantum computers
- 目前该开源代码中使用的求解工具为pyqubo,neal 包,可使用玻色量子开发的kaiwu SDK量子开发套件进行替换,请见项目文档
- 欢迎关注开物量子开发者社区,定期发布最新文章