广州免费建站排行视频号推广
生信碱移
空间多组学分析
SpatialGlue 可整合来自同一组织切片的多种空间组学模态,提升组织空间域的解析分辨率。其通过图神经网络为每种组学学习低维嵌入表示,并通过双重注意力聚合机制整合空间信息与多组学特征,自适应捕捉模态重要性,实现更精确的空间多组学整合分析。
空间转录组学是继单细胞转录组学之后的一项重大技术革新,能够实现空间级别的组学和分辨率。目前,越来越多的技术实现了在同一个组织切片上同时分析不同组学。空间技术主要可以划分为测序技术和成像技术两大类别。①测序技术包括DBiT-seq、spatial-CITE-seq、spatial ATAC–RNA-seq、CUT&Tag-RNA-seq、SPOTS、SM-Omics、Stereo-CITE-seq、spatial RNA-TCR-seq和10x Genomics Xenium等; ②成像技术包括DNA seqFISH+、DNA-MERFISH-based DNA和RNA profiling、MERSCOPE和Nanostring CosMx等。
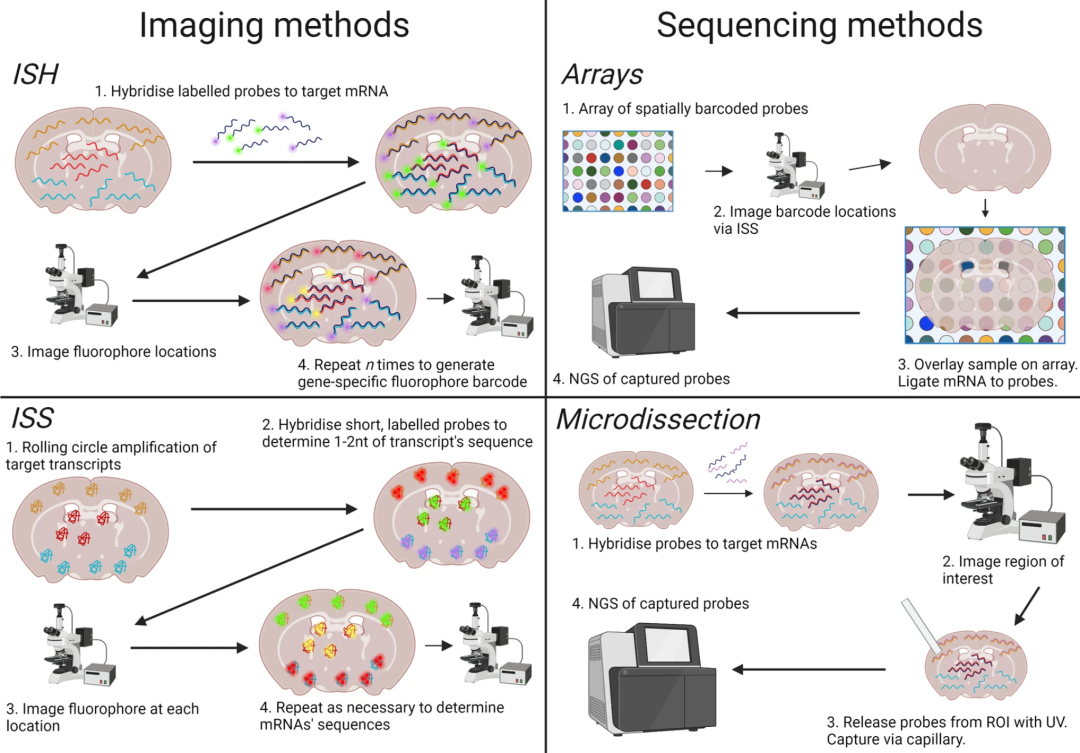
▲ 空间转录组技术的主要分类与原理概述。空间转录组技术主要分为两大类:①成像类技术直接在组织切片中对 mRNA 进行原位成像(图左),常见方法包括原位杂交(ISH)和原位测序(ISS)。ISH通过荧光标记的基因特异性探针与mRNA杂交,并通过多轮成像构建基因条形码;ISS则在组织中对扩增的转录本进行测序以识别其序列。②测序类技术通过提取组织中的 mRNA (图右),并在提取前记录其空间信息,再通过高通量测序进行基因鉴定。常见方式包括利用空间条码探针的阵列(array-based)或微切技术(microdissection-based)来标记空间位置信息。不同技术在空间分辨率、基因覆盖度及实验通量等方面存在权衡,适用于不同的组织尺度与研究需求。图中展示了这四类方法对转录本空间信息与物种信息的记录方式。DOI:10.1186/s13073-022-01075-1。
尽管目前存在多种技术手段,但多组学整合面临显著挑战。首先,①不同组学的特征维度差异巨大(如蛋白质与转录本数量),统计分布亦各异。同时,②整合跨组学特征与空间信息进一步增加难度,尚无专为同一组织切片获得的空间多组学数据设计的工具。非空间多组学整合方法包括Seurat WNN、MOFA+、StabMap、totalVI、MultiVI与scMM等。③但多数针对特定模态,适用性有限(如totalVI针对CITE-seq的RNA与蛋白,MultiVI适用于基因表达与染色质可及性)。④现有空间组学工具如 STAGATE、SpaGCN与GraphST,仅支持单组学,处理多组学数据时仅做特征拼接,未区分不同组学特征的重要性。因此,亟需专门面向空间多组学的工具,实现空间感知的跨组学整合。

▲ DOI: 10.1038/s41592-024-02316-4。
为此,来自新加坡国立大学的jinmiao chen研究团队提出了 SpatialGlue,一种面向空间多组学的整合方法,于2024年6月21号发表于Nature Methods [IF: 36.1]。SpatialGlue可整合来自同一组织切片的多种空间组学模态,提升组织空间域的解析分辨率。其通过图神经网络为每种组学学习低维嵌入表示,并通过双重注意力聚合机制整合空间信息与多组学特征,自适应捕捉模态重要性,实现更精确的多组学信息整合。作者在模拟数据及人类淋巴结实测数据上评估SpatialGlue性能,结果优于现有方法,并能识别更多解剖细节。

▲ SpatialGlue 方法概述。a. SpatialGlue 旨在同时整合不同组学模态与空间信息,从而获得对特定组织的全面分子表征。 b. SpatialGlue 模型结构首先利用 K 近邻算法,基于空间坐标构建空间邻接图,并基于归一化表达数据为每种组学模态构建特征邻接图。随后,对于每个模态,图神经网络编码器以归一化表达值和邻接图为输入,通过迭代聚合邻居节点的表示,学习两种图特异性表示。为了捕捉不同图结构的重要性,作者设计了组学内注意力聚合层,用于自适应整合图特异性表示并生成模态特异性表示。最后,为保留不同组学模态的重要性,SpatialGlue 采用组学间注意力聚合层,自适应整合模态特异性表示,输出最终的整合空间位点表示。
该工具的github链接如下,本文简要介绍淋巴结如何运用 SpatialGlue 整合人类淋巴结10x Genomics Visium 转录组与蛋白质多组学数据,更多分析方向可以自行学习。
-
https://github.com/JinmiaoChenLab/SpatialGlue
-
本文示例数据下载链接:https://drive.google.com/drive/folders/1RlU3JmHg_LZM1d-o6QORvykYPoulWWMI
0.软件安装
SpatialGlue基于python解释器,可以使用以下代码安装该库:
pip install SpatialGlue
除此之外,需要以下依赖库与软件:
- python==3.8
- torch>=1.8.0
- cudnn>=10.2
- numpy==1.22.3
- scanpy==1.9.1
- anndata==0.8.0
- rpy2==3.4.1
- pandas==1.4.2
- scipy==1.8.1
- scikit-learn==1.1.1
- scikit-misc==0.2.0
- tqdm==4.64.0
- matplotlib==3.4.2
- R==4.0.3
1.转录/蛋白组空间整合
① 导入软件,设置使用cpu/cuda与软件路径:
import os
import torch
import pandas as pd
import scanpy as scimport SpatialGluedevice = torch.device('cuda:2' if torch.cuda.is_available() else 'cpu')
os.environ['R_HOME'] = '/scbio4/tools/R/R-4.0.3_openblas/R-4.0.3'0
② 加载模态数据:
# 下载的数据路径
file_fold = '/input_dir'# 读取数据
adata_omics1 = sc.read_h5ad(file_fold + 'adata_RNA.h5ad') # 转录组
adata_omics2 = sc.read_h5ad(file_fold + 'adata_ADT.h5ad') # 蛋白adata_omics1.var_names_make_unique()
adata_omics2.var_names_make_unique()# 设置数据类型
data_type = '10x'# 设置随机数种子
from SpatialGlue.preprocess import fix_seed
random_seed = 2022
fix_seed(random_seed)
③ 数据预处理。SpatialGlue采用标准的转录组和蛋白质数据预处理步骤。具体而言,对于转录组数据,基因表达计数通过 scanpy 包进行对数转换和文库大小归一化处理。选取前3000个高变基因(HVGs)作为PCA降维的输入。为确保与蛋白质数据输入维度一致,保留前k个(蛋白质数量)主成分作为模型输入。蛋白质表达计数使用中心对数比(CLR)进行归一化。PCA 降维后的所有主成分均作为模型输入。
from SpatialGlue.preprocess import clr_normalize_each_cell, pca# 使用转录组模态的高变基因做整合
sc.pp.filter_genes(adata_omics1, min_cells=10)
sc.pp.highly_variable_genes(adata_omics1, flavor="seurat_v3", n_top_genes=3000)
sc.pp.normalize_total(adata_omics1, target_sum=1e4)
sc.pp.log1p(adata_omics1)
sc.pp.scale(adata_omics1)
#
adata_omics1_high = adata_omics1[:, adata_omics1.var['highly_variable']]
adata_omics1.obsm['feat'] = pca(adata_omics1_high, n_comps=adata_omics2.n_vars-1)# 蛋白模态
adata_omics2 = clr_normalize_each_cell(adata_omics2)
sc.pp.scale(adata_omics2)
adata_omics2.obsm['feat'] = pca(adata_omics2, n_comps=adata_omics2.n_vars-1)
随后,基于空间信息构建邻接图:
from SpatialGlue.preprocess import construct_neighbor_graph
data = construct_neighbor_graph(adata_omics1, adata_omics2, datatype=data_type)
④ 执行SpatialGlue模型。SpatialGlue模型通过自适应整合多个组学模态的表达信息,并结合空间位置信息,学习联合的潜在表示(代替传统scanpy/seurat分析的主成分PC)。模型训练后输出的结果包括 ①各组学模态的与其融合后的联合表示(可用于聚类、可视化和差异表达分析),以及②模态内与模态间的注意力权重(用于解释组学模态的相对贡献)。
# 初始化模型
from SpatialGlue.SpatialGlue_pyG import Train_SpatialGlue
model = Train_SpatialGlue(data, datatype=data_type, device=device)# 模型训练
output = model.train()# 整合模型embedding
adata = adata_omics1.copy()
adata.obsm['emb_latent_omics1'] = output['emb_latent_omics1'].copy()
adata.obsm['emb_latent_omics2'] = output['emb_latent_omics2'].copy()
adata.obsm['SpatialGlue'] = output['SpatialGlue'].copy()
adata.obsm['alpha'] = output['alpha']
adata.obsm['alpha_omics1'] = output['alpha_omics1']
adata.obsm['alpha_omics2'] = output['alpha_omics2']
模型训练完成后,SpatialGlue会返回包含多种输出结果的output文件,如上代码所示:
-
emb_latent_omics1:第一种组学模态的潜在表征 (embedding)。 -
emb_latent_omics2:第二种组学模态的潜在表征。 -
alpha_omics1:针对第一种组学模态的模态内注意力权重,解释每个图对每个簇的贡献。 -
alpha_omics2:针对第二种组学模态的模态内注意力权重,解释每个图对每个簇的贡献。
⑤ 下游分析示例,多模态空间融合表征的空间域鉴定。注意:下面用的是mclustr工具(需要在python中调用R语言),环境可能比较难配置,推荐还是使用常规的leiden或者louvain方法(常规scanpy/seurat流程的聚类方法):
# 使用mclust进行聚类,也可以选择常规的leiden或者louvain
from SpatialGlue.utils import clustering
tool = 'mclust' # mclust, leiden, and louvain
clustering(adata, key='SpatialGlue', add_key='SpatialGlue', n_clusters=6, method=tool, use_pca=True)
# 降维结果可视化
import matplotlib.pyplot as plt
fig, ax_list = plt.subplots(1, 2, figsize=(7, 3))
sc.pp.neighbors(adata, use_rep='SpatialGlue', n_neighbors=10)
sc.tl.umap(adata)
sc.pl.umap(adata, color='SpatialGlue', ax=ax_list[0], title='SpatialGlue', s=20, show=False)
sc.pl.embedding(adata, basis='spatial', color='SpatialGlue', ax=ax_list[1], title='SpatialGlue', s=25, show=False)
plt.tight_layout(w_pad=0.3)
plt.show()

▲ 基于多组学的空间域鉴定结果。
⑥ 看一下不同模态的重要性:
import pandas as pd
import seaborn as sns
plt.rcParams['figure.figsize'] = (5,3)
df = pd.DataFrame(columns=['RNA', 'protein', 'label'])
df['RNA'], df['protein'] = adata.obsm['alpha'][:, 0], adata.obsm['alpha'][:, 1]
df['label'] = adata.obs['SpatialGlue'].values
df = df.set_index('label').stack().reset_index()
df.columns = ['label_SpatialGlue', 'Modality', 'Weight value']
ax = sns.violinplot(data=df, x='label_SpatialGlue', y='Weight value', hue="Modality",split=True, inner="quart", linewidth=1, show=False)
ax.set_title('RNA vs protein')
ax.set_xlabel('SpatialGlue label')
ax.legend(bbox_to_anchor=(1.4, 1.01), loc='upper right')plt.tight_layout(w_pad=0.05)
#plt.show()

▲ 不同模态的重要性,可以看到attention在大部分聚类簇中更加关注蛋白质模态而非RNA。
简单分享到这里
各位佬哥佬姐试试哇